Four common pure fluids were chosen to elucidate the reliability of reactive force fields in estimating bulk properties of selected molecular systems: CH4, H2O, CO2 and H2. The pure fluids are not expected to undergo chemical reactions at... more
An accurate gravimetric method was used to explore water adsorption/desorption isotherms between 105 and to 250°C for a number of synthetic and natural porous solids including controlled pore glass, activated carbon fiber monoliths,... more
Quantum mechanical, molecular mechanics and molecular dynamics (MD) methods were used to investigate initial steps of 2 0 -deoxyuridine-5 0 -monophosphate (dUMP) methylation catalysed by the thymidylate synthase (TS) enzyme. The amino... more
Quantum mechanical, molecular mechanics and molecular dynamics (MD) methods were used to investigate initial steps of 2 0 -deoxyuridine-5 0 -monophosphate (dUMP) methylation catalysed by the thymidylate synthase (TS) enzyme. The amino... more
The effects of the electric field on the vapor-liquid equilibria of methanol and ethanol confined in a graphitic slit pore of width 4 nm using molecular dynamics simulations are reported. The vapor-liquid critical temperature of methanol... more
Multiscale modeling approach is proposed to characterize interfacial behavior of crosslinked epoxy nanocomposites. In this study, crosslinked epoxy composed of epoxy resin EPON 862 and curing agent TETA with varying crosslink densities... more
Processes using supercritical fluids constitute another special separation process option other than the membrane processes which have been presented in one of the previous issues as alternative to conventional separation processes.... more
A peptide mimotope of the CD52 antigen with the sequence T 1 SSPSAD 7 has been co-crystallised with the CAMPATH-1H antibody. Molecular dynamics (MD) simulations in explicit water of the T 1 SSPSAD 7 peptide in both antibody free and bound... more
Incorrect folding of proteins in living cells may lead to malfunctioning of the cell machinery. To prevent such cellular disasters from happening, all cells contain molecular chaperones that assist nonnative proteins in folding into the... more
Liquid crystals are synthetic and biological viscoelastic anisotropic soft matter materials that combine liquid fluidity with crystal anisotropy and find use in optical devices, sensor/actuators, lubrication, super-fibers. Frequently... more
We propose a concept for a homogenous computational model in carrying out cross-scale numerical experiments on liquids. The model employs the particle paradigm and comprises three types of simulation techniques: molecular dynamics (MD),... more
In the paper we make a short overview of computer models based on particle approach, which can be suitable for the simulation of fluid flow through porous media. We concentrate on Molecular Dynamics (MD) and Dissipative Particle Dynamics... more
We examine the utility of the virial equation of state (VEOS) for the prediction of the solubility of hexane in supercritical carbon dioxide. We computed virial coefficients up to fourth-order for this mixture at 353.15 K, using... more
Although recent experimental studies have demonstrated that doping of nanoporous carbons with nitrogen is an effective strategy for highly diluted formaldehyde capture, the impact of carbon surface chemistry and the pore size on... more
Generalitat Valenciana, Universidad de Alicante, Fisher, Top Industrie, Carburos Metalicos, Sitec, Air Products
Introduction 495 II. Methods of molecular modelling of proteins 495 III. Molecular modelling of different types of biological receptors 502 { This review completes the series of publications illustrating the main avenues of medicinal... more
Se analizó la marcha usando datos de posiciones (x, y) de marcadores en cadera, rodilla y tobillo. Se calcularon ángulos articulares para evaluar la movilidad y coordinación del movimiento. La velocidad y aceleración angular se estimaron... more
Ion channels are gated, i.e. they can switch conformation between a closed and an open state. Molecular dynamics simulations may be used to study the conformational dynamics of ion channels and of simple channel models. Simulations on... more
Despite the complexity of ion-channels, MD simulations based on realistic all-atom models have become a powerful technique for providing accurate descriptions of the structure and dynamics of these systems, complementing and reinforcing... more
Interfacial layers of both vapour/liquid and crystal/liquid aqueous solutions of sodium and caesium halides have been analysed in detail using four different methods for the identification of interfacial molecules and layers, and both... more
Taylor & Francis makes every effort to ensure the accuracy of all the information (the "Content") contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or... more
An algorithm is developed that finds the optimal orientation of a rigid molecular structure, represented by Nreference sites, with respect to the same number of sites in an observed structure. The optimal orientation is found by... more
An extension of Wertheim’s first-order thermodynamic perturbation theory is proposed to describe the global phase behavior of linear rigid tangent hard sphere chains. The extension is based on a scaling proposed recently by Vega and... more
Purpose. Cyclodextrins are known to be good solubility enhancers for several drugs, improving bioavailability when incorporated in pharmaceutical formulations. In this work we intend to assess and characterize the formation of inclusion... more
We present a 250ns simulation of the wild-type, biotin-liganded streptavidin tetramer in the solution phase and compare the trajectory to two previously published simulations of the protein in its crystal lattice. By performing both types... more
Here, we report the 1.53-Å crystal structure of the enzyme 7-cyano-7-deazaguanine reductase (QueF) from Vibrio cholerae, which is responsible for the complete reduction of a nitrile (C≡N) bond to a primary amine (H 2 C-NH 2 ). At present,... more
The main purpose of this work is to present a free-volume combinatorial term in predicting vapor-liquid equilibrium (VLE) and solid-liquid equilibrium (SLE) of polymer/solvent and light and heavy hydrocarbon/hydrocarbon mixtures. The... more
We present a joint Molecular Dynamics (MD) / Kinetic Monte-Carlo (KMC) study aiming at the atomistic description of charge transport in stacks of liquid-crystalline tetra alkoxy-substituted metal-free phthalocyanines. The molecular... more
The success of process design in chemical engineering and energy technology depends on the availability and accuracy of thermodynamic properties. In recent years, molecular modeling and simulation has become a promising tool to accurately... more
Density, self-diffusion coefficient, and shear viscosity of pure liquid water are predicted for temperatures between 280 and 373 K by molecular dynamics simulation and the Green–Kubo method. Four different rigid nonpolarizable water... more
The modelling of thermodynamic properties of liquids from local density fluctuations is relevant to many chemical and biological processes. The Kirkwood-Buff (KB) theory connects the microscopic structure of isotropic liquids with... more
We study the dissociation of hydrogen from molecule to atoms and show how we can compute thermodynamic and transport properties of both species in a mixture under non-ideal conditions. The small system method can be used to sample... more
The modelling of thermodynamic properties of liquids from local density fluctuations is relevant to many chemical and biological processes. The Kirkwood-Buff (KB) theory connects the microscopic structure of isotropic liquids with... more
We study the dissociation of hydrogen from molecule to atoms and show how we can compute thermodynamic and transport properties of both species in a mixture under non-ideal conditions. The small system method can be used to sample... more
In this study, the adsorption capacity of single-wall carbon nanotubes (SWCNTs) bundles with regard to the pure CH 4 , N 2 , CO and CO 2 gases at 298 K and pressure range from 0.01 up to 2.0 MPa has been investigated experimentally and... more
A novel multibinding species has been obtained by attaching four aliphatic polyamine chains to an iron(II)-polyimine centre, derived from 2,6-diacetylpyridine. Molecular simulations for the complex corroborate the evidence from 1H NMR... more
Defect production and amorphization due to energetic uranium recoils in zircon (ZrSiO 4 ), which is a promising ceramic nuclear waste form, is studied using molecular dynamics simulations with a partial charge model. An algorithm that... more
We performed all-atom molecular dynamics simulations for bulk cyclohexane and analysed the short-and medium-range structures in supercooled and glassy states by using the Voronoi tessellation technique. From the analyses of both the... more
Influence of Additives on Nucleation of Vanillin: Experiments and Introductory Molecular Simulations
Nucleation of vanillin (VAN) in 2-propanol/water in the presence of additives, viz., acetovanillone (AVA), ethyl vanillin (EVA), guaiacol (GUA), guaethol (GUE), 4-hydroxyacetophenone (HAP), 4-hydroxybenzaldehyde (HBA), and vanillic acid... more
Influence of Additives on Nucleation of Vanillin: Experiments and Introductory Molecular Simulations
Nucleation of vanillin (VAN) in 2-propanol/water in the presence of additives, viz., acetovanillone (AVA), ethyl vanillin (EVA), guaiacol (GUA), guaethol (GUE), 4-hydroxyacetophenone (HAP), 4-hydroxybenzaldehyde (HBA), and vanillic acid... more
An out-of-equilibrium simulation method for tracking the time evolution of glassy systems (or any other systems that can be described by hopping dynamics over a network of discrete states) is presented.
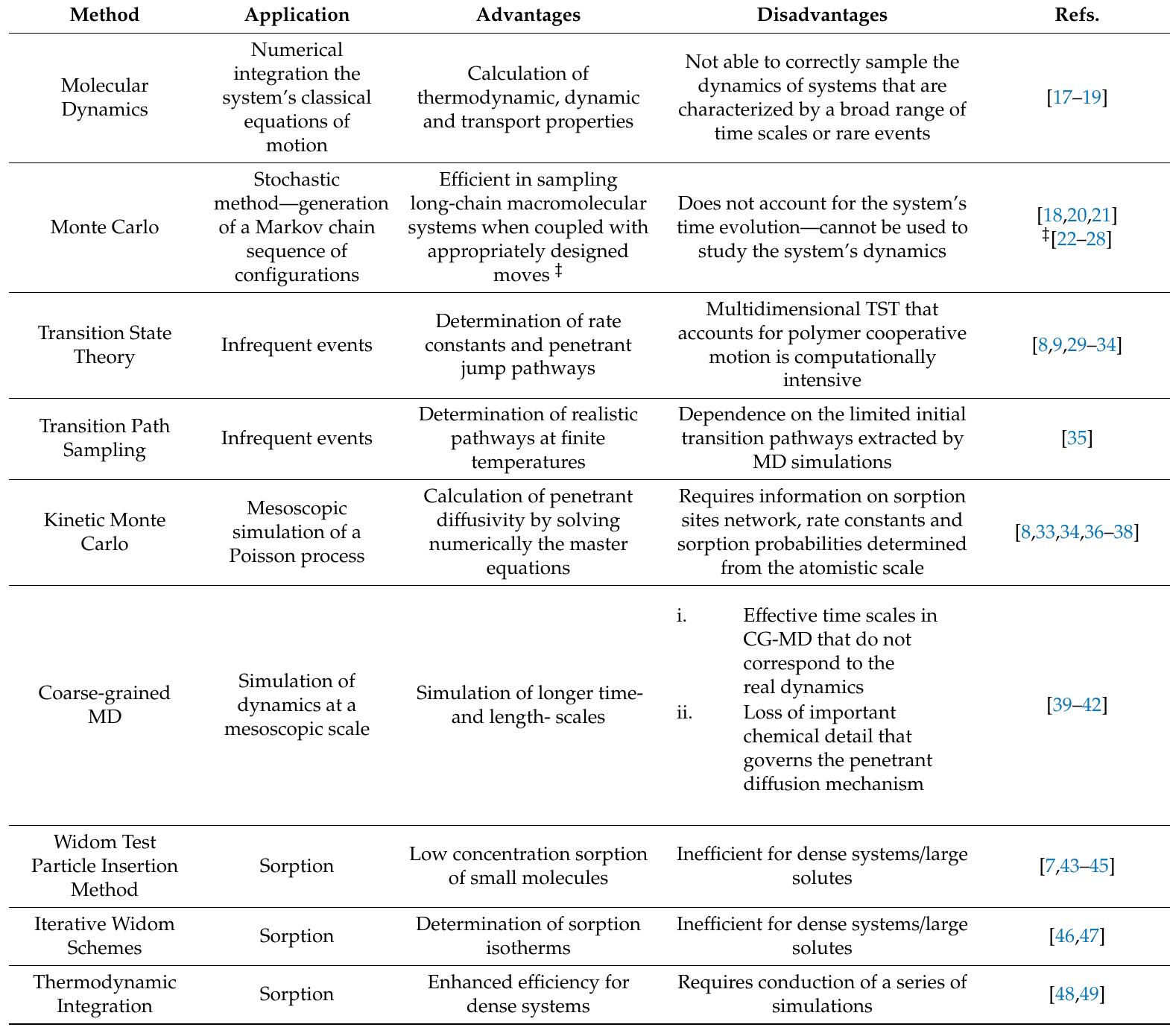
With a wide range of applications, from energy and environmental engineering, such as in gas separations and water purification, to biomedical engineering and packaging, glassy polymeric materials remain in the core of novel membrane and... more
![Figure 1. Molecular simulation methods at multiple length- and time scales. Hierarchical multiscale simulations utilize information extracted from the previous scale as input for conducting molecular simulations at longer length and time scales. Figure 1. Molecular simulation methods at multiple length- and time scales. Hierarchical multiscale The structure of the article is as follows: in Section 2 we present an introduction to basic concepts and discuss equilibrium and non-equilibrium molecular dynamics and Monte Carlo techniques for simulating polymers and sorption and diffusion of small molecules therein. Methodological aspects of the simulations are addressed very briefly—they have been presented extensively in earlier reviews. Emphasis is laid on multiscale methods that incorporate infrequent event analysis of elementary diffusive jumps and on coarse-graining/reverse mapping for the generation of realistic model configurations in which to study permeation phenomena. Both these aspects are necessary for predicting the separation performance of dense glassy polymer matrices, especially if their chemical constitution is complex. Section 3 discusses coarse-grained approaches to the simulation of sorption and diffusion phenomena in polymers per se. The approaches discussed have been designed to address long length and time scale phenomena by cutting down on the computational cost relative to molecular simulation algorithms is often too computationally demanding for such systems, there is increasing interest in multiscale modeling methods based on systematic coarse-graining of the molecular representation. For some categories of new membrane materials whose synthesis has taken off only recently, such as Metal Organic Frameworks, one sees simulation work going hand-in-hand with and even guiding, experimental efforts. High throughput computational screening for the identification of new nanoporous material structures that best satisfy the requirements of specific gas separation sequestration, or storage applications has emerged [10]. Machine learning techniques are starting to be used both in the development of more refined quantum mechanics-based classical force fields (or coarse grained ones based on the atomistic representation) for conducting simulations and in mining the copious data generated by molecular simulations for materials design.](https://figures.academia-assets.com/120703714/figure_001.jpg)
![Figure 2. Permselectivity performance of glassy polymers in comparison to rubbery polymers for O>/N>2 separation. Adapted with permission from [15]. A high performance separation membrane [13] is expected to exhibit high selectivity characterist combined, simultaneously, with satisfactory, or even, ideally, high permeability to one of the penetr. components. The selectivity upper bound. This empirical performance of membrane materials for gas separations is limited by imit originates from the fact than an increase in permeability of the mi permeable component is accompanied by a decrease in the separation ability. If P; is the permeabil of the more permeable gas, t he upper bound [13,14] is determined as a line in the log-log plot of 1 n relationship P; = Hr] , where n is the slope of the line, above which no data exist. Data near or j the upper bound correspond almost exclusively to glassy polymers and this is attributed prima1 to the higher solubility coefficients in glasses compared to rubbery polymers due to the increé of free volume below the glass transition temperature (T,), and in some cases partially to a bet diffusion selectivity of glassy polymers in comparison to the rubbery ones. In Figure 2, data selectivity for O2/N>2 separation are plotted as a function of O2 permeability for rubbery polymers a the upper bounds for both rubbery and glassy polymers [15,16] are shown that exhibit about an or¢ of magnitude difference in favour of the separation performance of glassy polymers.](https://figures.academia-assets.com/120703714/figure_002.jpg)

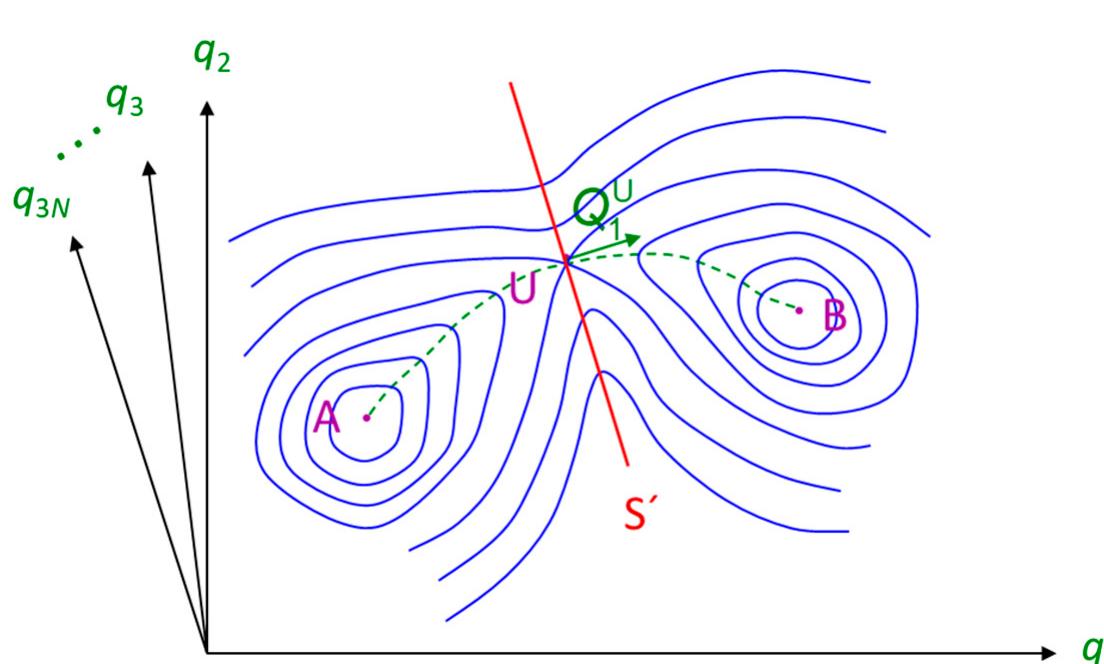
![Figure 5. Indicative narrow-necking regions in the accessible volume obtained from geometric analysis, which serve as initial estimates for the transition state search calculations [33]. The simulated polymer is a poly(amide imide) in a cubic box of edge length 28.09 Awith periodic boundary conditions. Atoms are not shown. The dark regions are clusters of accessible volume, as determined with a spherical probe of radius 1.1 A. Necking regions are identified by probing the same configuration with a smaller probe radius. Some of the necking regions are indicatively highlighted in yellow.](https://figures.academia-assets.com/120703714/figure_005.jpg)
![An ideal CG effective potential would enable the reproduction of target properties and wou 2 also transferable to other thermodynamic state points [137,138,148-151]. The CG model shou so allow, in a straightforward manner, reverse mapping [152-161] back to representative atomis: ynfigurations (usually on the basis of geometric criteria) of the well equilibrated structures at t G level. Special measures must be taken for macromolecular structures with complex chemic ynstitution to avoid potential overlaps or concatenations [162]. The final atomistic configuratio om reverse mapping are then subjected to a final relaxation of the system’s local interactions. Figure lustrates a hierarchical backmapping procedure used for the equilibration of polystyrene mode three different representations [161]. The multi-resolution method proposed in this study enabl ie generation of well-equilibrated high molecular weight polymer configurations that are initia! juilibrated at the largest length scale corresponding, in this case, to a soft-blob based representati ‘igure 6c). The detailed description of the system is introduced stagewise, first migrating from t ft-blob based to a moderately CG model (Figure 6b) and equilibrating the system locally at this lev id subsequently re-inserting the microscopic detail (Figure 6a) and equilibrating again at this sta, he hierarchical backmapping using a sequence of models aims at the efficient generation of realis ng chain polymer melts in a computationally feasible manner, preserving the long-waveleng iaracteristics of the macromolecular systems under study. Figure 6. Depiction of a hierarchical backmapping corresponding to (a) a united atom representation, (b) a moderate CG model consisting of beads of types A and B, and (c) a blob-based representation of soft spheres by lumping together a number of beads of representation (b). The backmapping procedure is described in (d) for the three resolutions. Reproduced with permission from [161].](https://figures.academia-assets.com/120703714/figure_006.jpg)
![a > ii: ae As a general rule, glassy polymers have a higher sorption capacity than rubbery polymers with the glassy sorption isotherms exhibiting a concave curvature in contrast to the rubbery one: hat are almost linear. During sorption at high concentrations of highly condensable penetrant: such as COz, the potential energy barriers that resulted in disjoint subsets of configurations ir he dense amorphous glass are altered, enabling a more frequent barrier crossing. This fact lead: o volume swelling phenomena that are observed both in glassy and rubbery polymers, due to < penetrant-induced increase in the fractional free volume and to plasticization of the dense amorphou: polymer matrix [175-180]. These phenomena directly influence the performance of the material as < separation medium [181], enhancing the permeability and diffusivity as the concentration increases The presence of the penetrant at high concentrations accelerates the segmental dynamics of the chains resulting in the case of polymer glasses in the reduction of the glass transition temperature due to polymer softening. Plasticization phenomena are described in terms of a characteristic pressure (known as “plasticization pressure”) which is related to a minimum in the gas permeability (Figure 7) Under those circumstances, sorption isotherms of glassy polymers at high pressures may exhibit < rubber type linear behavior, while following a glassy-type curve at lower pressures. Swelling anc plasticization behavior of a polymeric system is affected by temperature [182] and this can be considerec as an outcome of the interplay of the opposite trends between sorption capacity and chain mobility a: a function temperature. Figure 7. Experimental measurement of CO, permeability, solubility and diffusion coefficient as a function of pressure in a polyimide membrane. The black dashed-line arrow is indicative of the characteristic pressure (“plasticization pressure”) that corresponds to the minimum of permeability. Reproduced with permission from [183]. Figure 7. Experimental measurement of CO permeability, solubility and diffusion coefficient as a](https://figures.academia-assets.com/120703714/figure_007.jpg)
![Figure 8. Molecular simulation results on (a) isotherms of CO sorption and (b) polymer swelling for polystyrene at various temperatures and pressures. Reproduced with permission from [47]. volume was conducted to facilitate efficient loading of CO2. The polymer-solute matrices were subjected to NPT MD simulations during which the solute molecules were swapped between accessible volume cavities to achieve an efficient equilibrium repartitioning. The chemical potential calculations were performed using the Direct Particle Deletion method [47]. They also analyzed the polymer segmental relaxation times in the presence of CO» to obtain estimates of the solvent-induced glass transition pressure at various temperatures below the Tg of pure polystyrene, obtaining good agreement with available experimental measurements. Sorption and dilation effects have been modeled in fluorinated polyimides (6FDA-ODA,6FDA-DPX,6FDA-DAM) [183,192,193], in poly (ether sulfone) (PES) and polysulfone [185] polymers and in copolyimide polymers [188] up to high pressures. Performance and plasticization resistance of an emerging class of polymeric membrane materials called ‘thermally rearranged polymers’ [194—204] that are generated by thermally modifying aromatic polyimide chains, has also been investigated computationally [205-211] in recent years. These phenomena have been also simulated for high free volume polymers of intrinsic microporosity (PIM) [56,189,212,213].](https://figures.academia-assets.com/120703714/figure_008.jpg)
![Figure 9. (a) Average (dimensionless) solubility as a function of pressure for O2 and Nz ina 6FDA-6FpDA polyimide glassy polymer film excluding (dashed lines) or including (solid lines) interactions with the other air molecules. (b) Solubility selectivity for O2/N2 separation in the same film. Reprinted with permission from [223].](https://figures.academia-assets.com/120703714/figure_009.jpg)
Prokofiev for their time and effort in serving in my dissertation committee. I thank Prof. Maroudas for the opportunity to attend his group meetings which have always been a valuable learning experience. I would like to thank Prof.... more
Prokofiev for their time and effort in serving in my dissertation committee. I thank Prof. Maroudas for the opportunity to attend his group meetings which have always been a valuable learning experience. I would like to thank Prof.... more
Prokofiev for their time and effort in serving in my dissertation committee. I thank Prof. Maroudas for the opportunity to attend his group meetings which have always been a valuable learning experience. I would like to thank Prof.... more
Being HIV-1-PR an essential enzyme in the viral life cycle, its inhibition can control AIDS. Because the folding of single domain proteins, like HIV-1-PR is controlled by local elementary structures (LES, folding units stabilized by... more