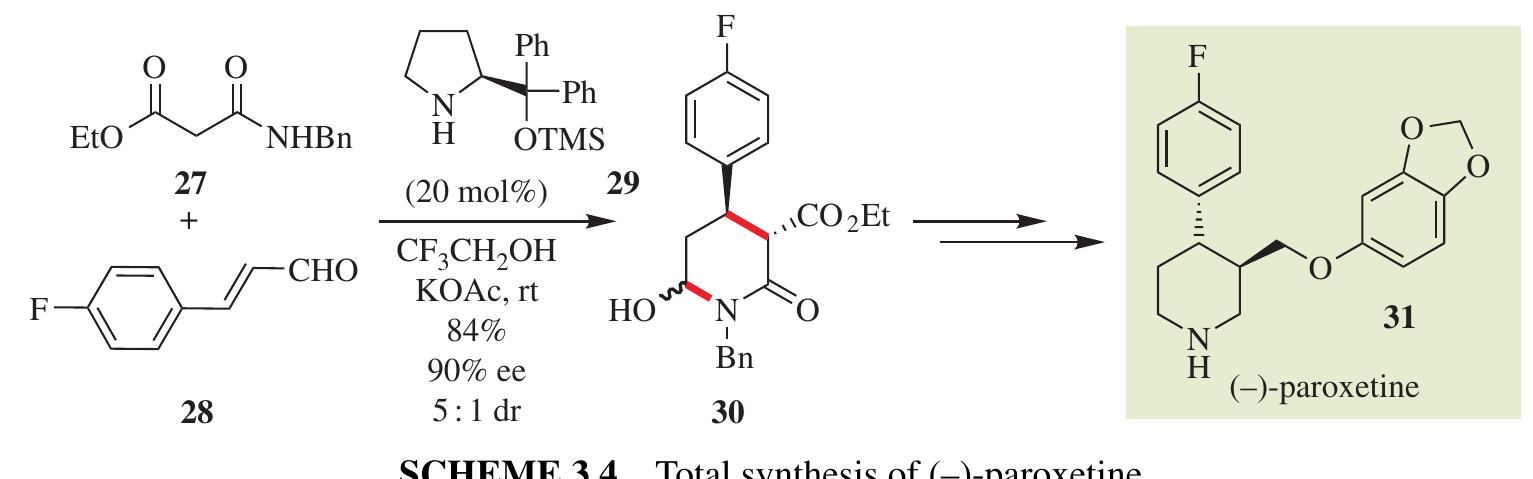
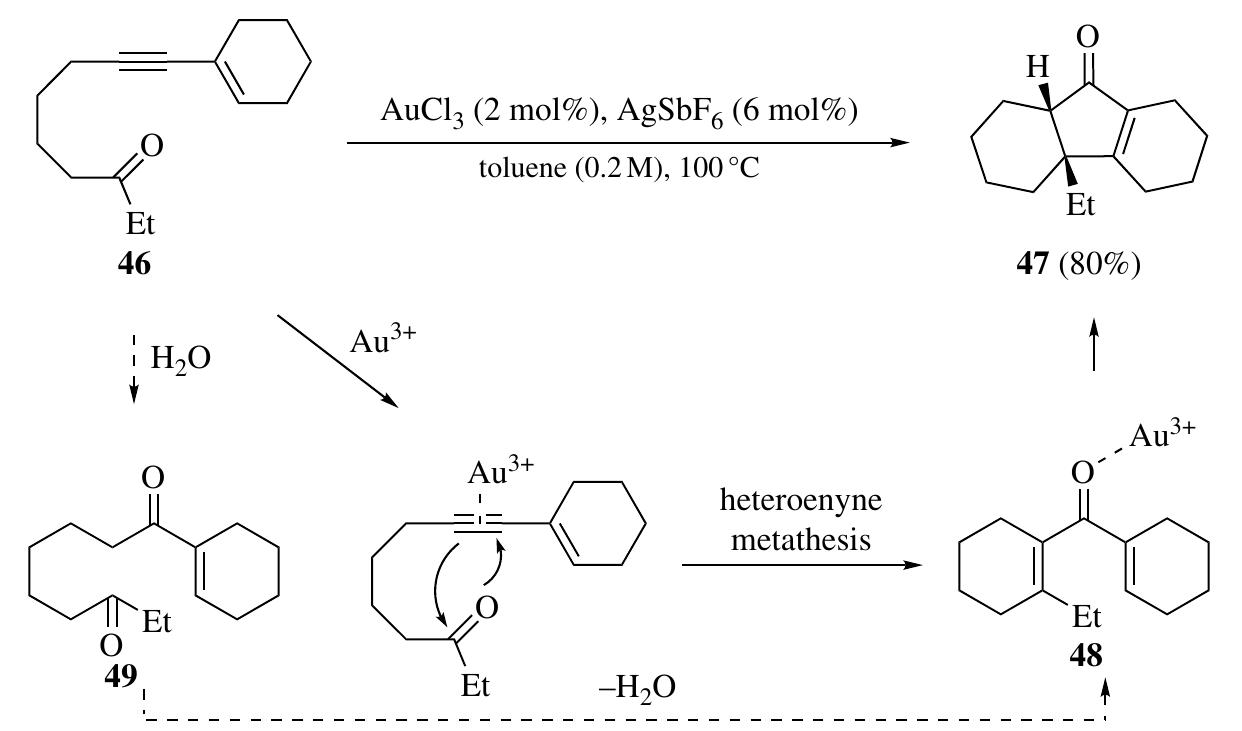
 Fang zhang
Fang zhangAI-generated Abstract
Chiral amine-mediated organocatalytic cascade reactions represent significant advancements in organic synthesis. This paper discusses the mechanisms underlying these reactions, particularly focusing on enamine and iminium activations. It highlights various cascade processes, demonstrates their applications, and underscores the impact of multiple catalyst systems in enhancing reaction efficiencies.
Figures (539)







![SCHEME 1.8 Two-step direct proline-catalyzed enantioselective synthesis of hexoses. To improve the efficiency and selectivity of the tandem aldol process, Cérdova’s group also isolated the B-hydroxyaldol intermediate from the first aldol transformation prior to the second aldol reaction. The pure intermediate was subjected to the second aldol reaction with a different catalyst (Scheme 1.8). The two-step synthetic protocol made it possible to investigate both (L)- and (D)-catalysts in stereocontrol. The synthesis of hexoses proceeded with excellent chemo-, diastereo-, and enantioselectivity. In all cases except one, the corresponding hexoses were isolated as single diastereomers with >99% ee [10]. 1.2.1.3 Enamine-Enamine in Three-Component Cascades As part of acontinuing effort, Chowdari et al. reported L-proline-catalyzed direct asymmetric assembly reactions involving three different components—aldehydes, ketones, and azodicarbox- ylic acid esters—to provide optically active functionalized B-amino alcohols in an enzyme-like fashion. These are the first examples of using both aldehydes and ketones as donors in one pot (Scheme 1.9) [11].](https://figures.academia-assets.com/35916932/figure_008.jpg)
![Moreover, enamine catalytic in situ sequences of acetaldehyde with two electrophiles can be envisioned (Scheme 1.11). The first successful realization of this concept with a proline-catalyzed double Mannich reaction of acetaldehyde with N-Boc-imines 36 was developed to give pseudo-C,-symmetric B,B’-diaminoaldehydes 37 with extremely high stereoselectivities (>99:1 dr, >99% ee) [13]. A similar approach with ketones was also realized [14].](https://figures.academia-assets.com/35916932/figure_009.jpg)



![SCHEME 1.13 Design of an enamine—iminium cascade with enones. a eet CR A Rs In addition to the consecutive aldol reactions of aldehydes, Barbas’s group also eported enamine-activated Diels-Alder reactions (or double Michael reactions) yetween o,B-unsaturated ketones and nitroolefin (Scheme 1.15) for the first time in 2002 [17]. In contrast to MacMillan’s iminium catalysis for Diels—Alder reactions, wherein o,f-unsaturated carbonyl compounds were activated as dienophiles in a ,UMO-lowering strategy based on iminium formation [3], an alternative strategy nvolving the in situ generation of 2-amino-1,3-dienes from o.,B-unsaturated ketones](https://figures.academia-assets.com/35916932/figure_013.jpg)










![diethyl azodicarboxylate [33], Han et al. extended inverse-electron-demand aza-Diels— Alder reaction of electron-deficient N-sulfony1-1-aza-1,3-butadienes to o1,8-unsaturated aldehydes to construct chiral piperidine derivatives bearing several functional groups in a straightforward manner (Scheme 1.27) [34]. Moderate to good yields (66 to 95%), good diastereoselectivities (E/Z=8 : 1), and excellent enantioselectivities (97 to 99% ee) were observed for this system.](https://figures.academia-assets.com/35916932/figure_024.jpg)
![The asymmetric inverse-electron-demand aza-Diels—Alder reaction of N-Ts-1- aza-1,3-butadienes derived from 3-argiocarbonylcoumarins and acetaldehyde has also been developed using chiral aminocatalysis, giving tricyclic chroman-2-one derivatives in high e1 enantioselectivities $ (up to 95% b ce) [35]. a ee “* ee rr rr ee a a ae](https://figures.academia-assets.com/35916932/figure_025.jpg)



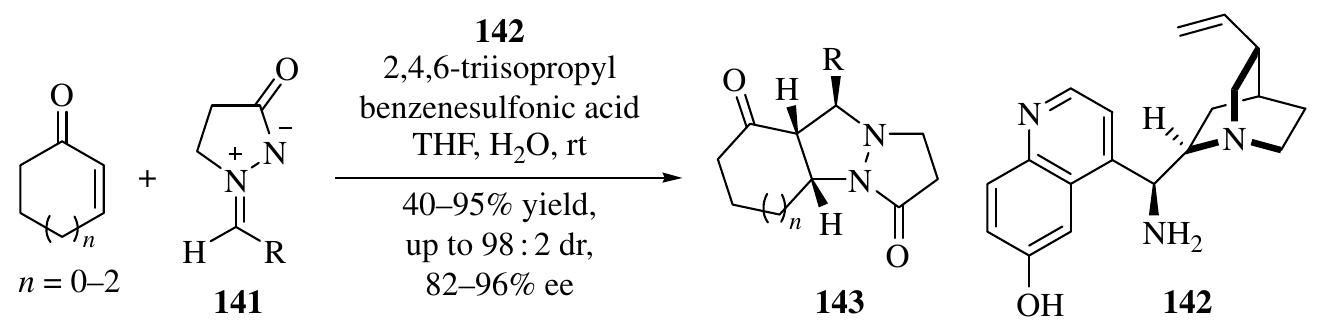
![a diethyl vinylphosphonate derivative [44], multisubstituted structurally diverse cyclo- hexene carbaldehydes with several stereogenic centers were efficiently synthesized. Enders et al. also developed an efficient one-pot procedure that provided direct entry to diastereo- and enantiomerically pure (299% de, ee) polyfunctionalized tricyclic frame- works 95 [45] (Scheme 1.32). The organocatalytic triple cascade, followed by a Diels— Alder sequence, leads to decahydroacenaphthylene and decahydrophenalene cores. Was ALY Lealsey [Ty]. In an effort to develop new cascade reactions, Zhang et al. envisioned that a linear alde- hyde can also be generated in situ via an extra iminium catalysis from an o1,B-unsaturated aldehyde prior to the triple cascade reaction. Therefore, there would be a possibility of extending the triple cascade reactions to four-component cascade reactions. Based on this design, a four-component quadruple cascade reaction through iminium—enamine— iminium-—enamine sequential activation initiated by oxa-Michael addition of alcohol to acrolein in moderate yield (about 50%), excellent diastereoselectivities (>20:1), and excellent enantioselectivities (>99% ee) was accomplished (Scheme 1.33) [47].](https://figures.academia-assets.com/35916932/figure_030.jpg)
![A similar organocatalytic quadruple domino Friedel—Crafts/Michael/Michael/ aldol condensation reaction initiated by Friedel-Crafts reaction of indole to acrolein was also developed by Enders et al. [48], as well as a microwave-assisted quadruple cascade organocatalytic Michael/Henry condensation/Michael/aldol condensation employing acetaldehyde and nitroalkenes as substrates [49].](https://figures.academia-assets.com/35916932/figure_031.jpg)
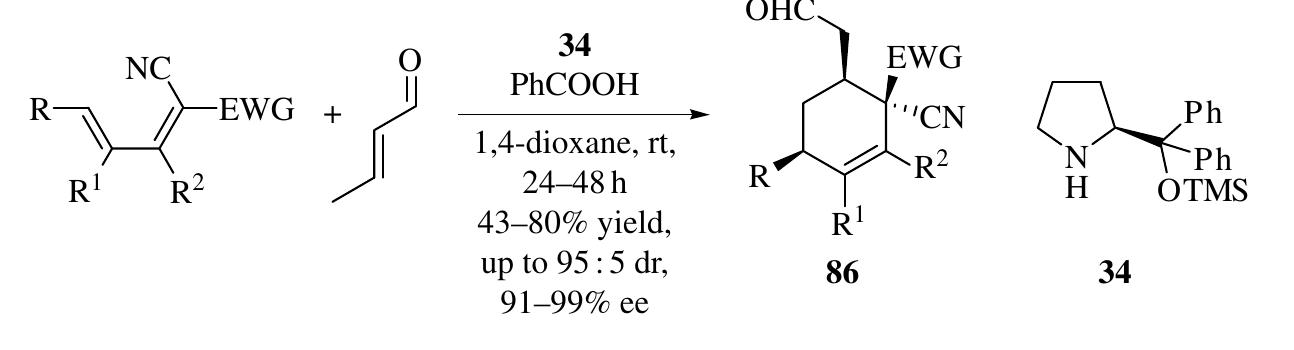
![1.2.3.1 Design of Enamine-Cyclization Cascade Reactions The nucleophilic Y in intermediate 6 can react with other electrophiles intermolecularly (Scheme 1.34a) or intramolecularly (Scheme 1.34b) as well as with the iminium ion. Moreover, the car- bonyl group of 6 can also undergo intramolecular aldol reaction with nucleophilic X (Scheme 1.34c). These nucleophilic addition reactions after enamine catalysis induce cyclization reactions to produce versatile five- or six-membered ring structures. 2.3.2 Enamine-Intermolecular Addition Cascades It was suggested that th ntermediate y-nitroaldehyde 91 in Scheme 1.31 might react with an aldehyde via a yx0-Henry sequence, and subsequent hemiacetalization would provide tetrahydropy an derivatives. Uehara et al. [50] and Iskikawa et al. [51] realized this hypothesi ndependently through a four-component reaction in one pot to furnish highl ubstituted tetrahydropyran derivatives 102 with excellent diastereo- and enantioselec ivity (up to 98 : 2 dr and 99% ee) (Scheme 1.35). These two methods are complementar: yecause anti-Michael products were synthesized using catalyst 101 [50], while syn Vlichael products were obtained with diphenylprolinol silyl ether catalyst 34 [51]. A similar strategy was used in the synthesis of piperidine derivatives when th -nitroaldehydes 91 were reacted with an imine through a Henry reaction followe](https://figures.academia-assets.com/35916932/figure_032.jpg)





![SCHEME 1.42 Mechanism proposed for organocatalyzed Diels—Alder reactions. Northrup and MacMillan extended the iminium-mediated Diels—Alder reactions to «,B-unsaturated ketones using a new chiral amine catalyst (Scheme 1.43) [66]. They found that cycloaddition of o,f-unsaturated ketones was unsuccessful with the chiral amine salts previously identified as excellent catalysts for enal activation. In contrast, the 2-(5-methylfuryl)-derived imidazolidinone 118 afforded good levels of enantiofacial discrimination while maintaining high reaction efficiency (89% yield, 25:1 endo/exo, 90% ee).](https://figures.academia-assets.com/35916932/figure_038.jpg)

![It was difficult to activate a-branched aldehydes such as acroleins with secondary amines because of the steric effect of poor generation of the corresponding iminium ions. An enantioselective Diels—Alder reaction with o-substituted acroleins 124 was realized by a primary amine organocatalyst 125 (Scheme 1.45) [68]. Acyclic dienes](https://figures.academia-assets.com/35916932/figure_040.jpg)
![In addition to conventional Diels—Alder reactions, consecutive [4+2] reactions have been subjected to extensive investigation through the iminium—enamine catalytic sequence. Wang, Rios, and others simultaneously described enantioselective cascade sulfa-, oxa-, and aza-Michael/aldol/dehydration reactions promoted by chiral secondary amines. An initial strategy for a one-pot synthesis of chiral thiochromenes with good to high enantioselectivities was reported (Schemes 1.46 and 1.47) [71].](https://figures.academia-assets.com/35916932/figure_041.jpg)

![Carlone et al. assumed that reactants bearing 1,4-nucleophilic—electrophilic sites for sequential [4+2] reactions of enals could be possible (Scheme 1.49) [82]. The overall [3+2+ 1] reaction was thus achieved with 2 equiv enals and | equiv malono- nitrile to afford cyclohex-1-ene-carbaldehyde derivatives 134 in good to high yields and a nearly enantiopure diastereomer. Other nucleophilic carbon-initiated sequen- tial [4+2] reactions of «,f-unsaturated aldehydes were also accomplished [83].](https://figures.academia-assets.com/35916932/figure_043.jpg)
![SCHEME 1.49 Cascade reaction of enals with malononitrile. 1.3.4 Iminium-Activated [3+ 2] Reactions](https://figures.academia-assets.com/35916932/figure_044.jpg)








![1.3.6.1 Iminium-Activated Cyclopropanations Kunz and MacMillan develope a highly efficient protocol for the construction of enantioenriched cyclopropane using stabilized ylides with dihydroindole catalysts (Scheme 1.60) [100 2-Carboxylic acid dihydroindole 154 might function as a directed electrostati activation (DEA) cyclopropanation catalyst. Iminium 156 and the ylide 153 engage in electrostatic association via their pendant carboxylate and thionium substituent: The zwitterion 156 would predominately populate the (Z)-iminium isomer t minimize van der Waals interactions between the substrate olefin and the ary hydrogen. As a result, the carboxylate group on the catalyst framework would direc ylide addition selectively to the Re-face of the activated olefin, thereby ensurin snantiocontrol and facilitating carbon-carbon bond formation. A second-generation catalyst in which the carboxylic acid of (S)-(—)-indoline-2- carboxylic acid was replaced by tetrazolic acid was used to improve enantioselectiv- ity as a consequence of increased steric bulk while retaining important structural functionality associated with the proposed directed electrostatic activation mode [101]. Combination of the iminium catalysis with arsonium ylides also provided access to cyclopropanes with high enantioselectivity [102].](https://figures.academia-assets.com/35916932/figure_054.jpg)


![SCHEME 1.63 Organocatalytic epoxidation of enals with hydrogen peroxide. 1.3.6.2 Iminium-Activated Epoxidations Similar to ylide 153 and bromomalo- nates 157, it was proposed that hydrogen peroxide could also be used as an amphi- philic reactant for [2+1] reactions of o,B-unsaturated aldehydes to furnish epoxidation products. Inspired by this hypothesis, Jorgensen’s group developed an organocatalytic asymmetric epoxidation system of ,B-unsaturated aldehydes with H,O, as the oxidant (Scheme 1.63) [106]. The reactions take place under mild condi- tions in good to high yields and enantio- and diastereoselectivities.](https://figures.academia-assets.com/35916932/figure_057.jpg)
![Despite the excellent results of epoxidation of simple o,f-unsaturated aldehydes, a general method for the epoxidation of -branched o,B-unsaturated aldehydes was challenging. After several years, the process was realized by the combination of a chiral primary Cinchona-based amine and a chiral phosphoric acid as cocatalysts, making it possible to achieve high efficiency (Scheme 1.65) [107]. It is believed that chiral phosphoric acid provides additional enantiodiscrimination in both steps as a chiral counterion in 160a and as a Brgnsted acid in 160b. This is supported by the match or mismatch observed when the phosphoric acids (R)-TRIP and (S)-TRIP were used in parallel studies.](https://figures.academia-assets.com/35916932/figure_058.jpg)

![SCHEME 1.67 Catalytic cycle proposed for epoxidation of enones. NHauOl OL We fEPI-OULOAY alllOn. It was found that 4-substituted o,o-diarylprolinol 162 catalyzed the asymmetric »poxidation of a,B-enones to give the corresponding chiral epoxides in good yields ind high enantioselectivities (up to 96%) under mild reaction conditions Scheme 1.68) [109]. The extension of epoxidation to cyclic «,B-unsaturated ketones with chiral primary salts was reported by Wang et al. in good yields and excellent snantioselectivities (up to 99%) [110].](https://figures.academia-assets.com/35916932/figure_060.jpg)
![It is proposed that efficient shielding of the Si-face of the chiral iminium intermediate by the bulky aryl groups of the catalyst leads to a stereoselective Re- facial nucleophilic conjugate attack on the electrophilic B-carbon by the amino group of 163 (Scheme 1.70). Then the chiral enamine intermediate generated performs a 3-exo-tet nucleophilic attack on the now electrophilic nitrogen atom, and acetic acid is released. The intramolecular ring closure pushes the equilibrium in the forward direction and makes this step irreversible. Aziridinations of o,f-unsaturated ketones triggered by chiral primary amine salts via iminium catalysis were reported soon after (Scheme 1.71) [113]. The reduced steric constraint of primary amines offers the unique possibility of catalyzing processes between sterically demanding partners, overcoming the inherent difficulty of chiral secondary amine catalysis. The reaction affords valuable N-Cbz- as well as N-Boc-protected aziridines 167 with almost complete diastereocontrol and very high enantioselectivity (up to 99% ee).](https://figures.academia-assets.com/35916932/figure_061.jpg)


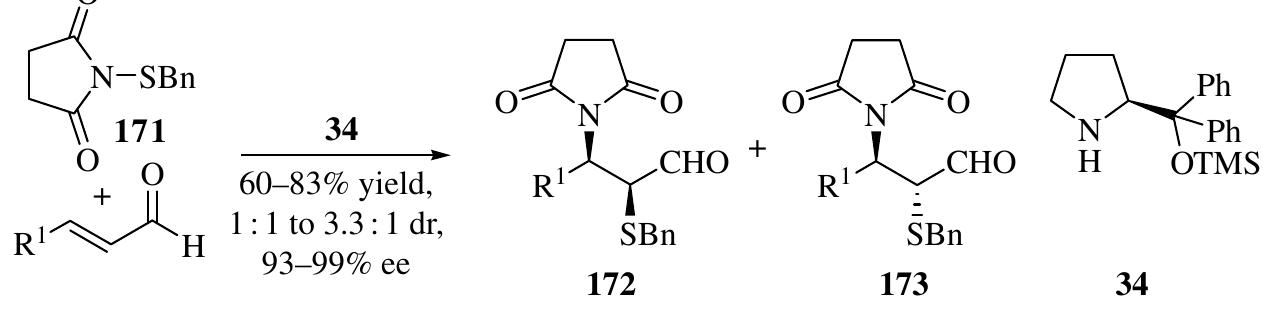
![An organocascade aminofluorination reaction of o.,B-unsaturated aldehydes witl 174 and NFSI (N-fluorobenzenesulfonimide) as an electrophilic fluorination agent was developed to produce chiral «-fluoro-B-amino aldehydes using catalyst 34 (Scheme 1.74) [116]. Up to 85% yield, 98 : 2 dr, and 99% ee of the reduced alcohols 175 were achieved Quintard and Alexakis developed a double Michael addition reaction of enals taking advantage of the high reactivity of vinyl sulfone—initiated nucleophilic addition of benzaldoxime, triazole, Angelica lactone, benzyl mercaptan, and 174 The powerful organocascade allows for the rapid construction of highly attractive synthons in high enantioselectivities (typically, 99% ee) [117]. 1.3.8 Iminium-Activated [3+3] Reactions](https://figures.academia-assets.com/35916932/figure_065.jpg)
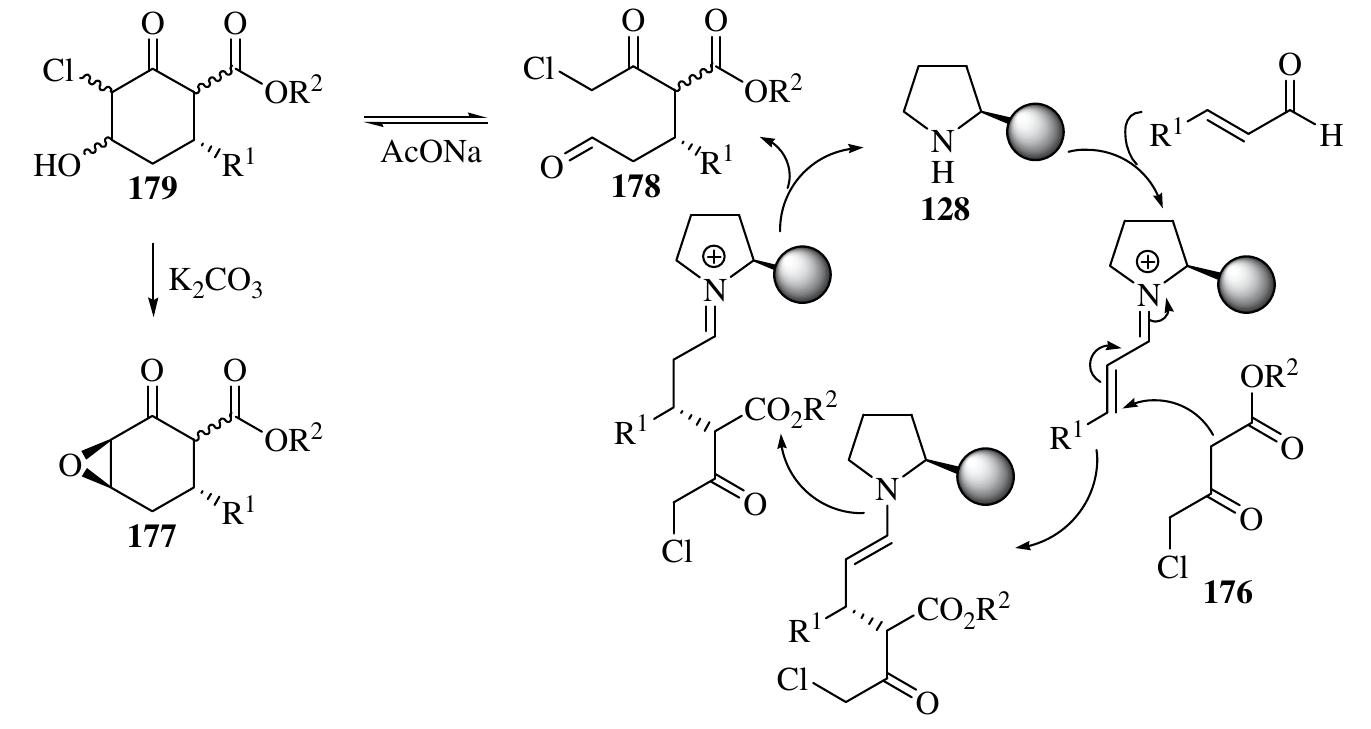
![In addition to the cyclization reactions above via the widely applied iminium— enamine sequence, in which diverse amphiphilic substrates bearing nucleophilic and electrophilic groups are added simultaneously to a,B-enals, [3+3] reactions can also be conducted by employing reactants bearing 1,3-nucleophilic sites in the iminium- mediated reactions of o,f-enals to furnish six-membered rings (Scheme 1.75). Reactants bearing 1,3-nucleophilic sites, such as enamines, enols, and 1,3-nucleophilic carbon species, have been used in iminium-activated [3+3] reactions. Furthermore, five-membered rings have also been synthesized through a similar approach with reactants bearing 1,2-nucleophilic sites, such as N-protected hydroxylamines [118]. 1.3.8.1 Iminium-Activated All-Carbon-Centered [3+3] Reactions The first highly enantioselective organocatalytic [3+3] reaction through Michael—Darzens condensation giving highly functionalized complex epoxycyclohexanone deriva- tives with up to four chiral centers was developed by Marigo et al. with excellent diastereo- and enantioselectivities (Scheme 1.76) [119]. The one-pot organocata- lytic domino reactions between y-chloro-B-keto esters 176 and o,f-unsaturated aldehydes occurred with catalyst 128 and AcONa as additive. The product was then](https://figures.academia-assets.com/35916932/figure_066.jpg)





![With regard to the mechanism, it was assumed that the reaction of diphenylprolinol ether 128 with o,f-unsaturated aldehyde resulted in an intermediary iminium ion (Scheme 1.83). Subsequent 1,4-addition of 2-hydroxy-1,4-naphthoquinone 189 to imin- ium ion followed by isomerization gives rise to the adduct 191. After hydrolysis, acetyliza- tion yields the desired 1,4-naphthoquinones 190 with regeneration of the catalyst. The equilibrium between 1,3-diones and the corresponding enol form renders it a suitable reactant for [3+3] reactions of o,B-unsaturated aldehydes [128]. The Michael—Morita—Baylis—Hillman reaction between o,f-unsaturated aldehydes and 5-substituted Nazarov reagent (Scheme 1.81) was inhibited because of the steric effect. However, oxo-[3 +3] cyclization proceeded in good yields and high enantiose- lectivies in this system [129]. La a) ee Le ee ee ee i ee i, cae ey i: fey ie rs , OT ... 2.4? ff 2. Qn](https://figures.academia-assets.com/35916932/figure_073.jpg)

![SCHEME 1.85 Catalytic enantioselective hydride-transferring closure. It is observed that 1,5-hydride transfer can be accelerated by iminium activation. Therefore, it is speculated that cinnamaldehyde derivatives 194 represent ideal acceptors that are susceptible to activation by secondary amine catalysts capable of forming an iminium ion (Scheme 1.85) [132]. The resulting iminium ion activation is expected to increase hydride transfer to alkene. The subsequent ring-closure reaction mediated by enamine catalysis furnishes ring-fused tetrahydroquinoline derivatives in moderate yields and high levels of enantioselectivity.](https://figures.academia-assets.com/35916932/figure_075.jpg)



![SCHEME 1.89 Aza-Michael/Mannich cascade by cycle-specific catalysis. queous or organic phase, was also developed by Fréchet’s group [138]. In addition to iminium-initiated cascade reactions, two of the steps in enamine- tivated cascade reactions can also be enforced by cycle-specific catalysis. It is well nown that diphenylprolinol silyl ether catalyst 34 is optimal for diverse enamine- nediated transformations to furnish products with high enantioselectivities. However, imilar to imidazolidinone catalysts, it proved to be less effective or ineffective for functional enamine catalysis. Cycle-specific catalysis via an aza-Michael/Mannich equence by combining 34 and either enantiomer of proline was thus developed to renerate 206 in about 60% yields with excellent diastereo- and enantioselectivities Scheme 1.89) [139].](https://figures.academia-assets.com/35916932/figure_079.jpg)














![Despite the great achievements that have been made in catalytic asymmetric 1,3-dipolar cycloadditions, electron-deficient carbon-carbon triple bonds had never been used as dipolarophiles until Shi et al. treated them with aldehydes 3 and amino esters 49 in the promotion of phosphoric acid 5e (Scheme 2.18). This reaction pro- vides an unprecedented approach to accessing 2,5-dihydropyrrole skeletons 63 in perfect enantioselectivities of up to >99% ee [29].](https://figures.academia-assets.com/35916932/figure_094.jpg)




![SCHEME 2.21 Phosphoric acid—catalyzed asymmetric synthesis of cyclic aminals. Rearrangement reactions are important carbon-carbon formation methods that enable rapid access to complex structures from simple starting materials, thus have great potential in the synthesis of biologically relevant molecules and natural products. In 2008, Rueping and Antonchick realized the first catalytic enantioselec- tive aza-Cope rearrangement reaction by employing phosphoric acid 6d as the catalyst, which provided an efficient route to optically active homoallylic amine derivatives 81 from aldehydes and diaryl homoallylic amines 80 (Scheme 2.23) [34a]. Later, Ren and Wulff successfully applied a vaulted biaryl ligand—derived chiral polyborate catalyst 82 to promote this aza-Cope rearrangement reaction, both aromatic and aliphatic aldehydes were good substrates to afford the desired amine products 8la with excellent enantioselectivities [34b]. Significantly, the addition of achiral benzoic acid led to a dramatic enhancement in the enantioselection, indi- cating a synergistic interaction of these two Brgnsted acids.](https://figures.academia-assets.com/35916932/figure_099.jpg)


![Jiang et al. proved that phosphoric acid can efficiently activate 1-aza-1,3-butadienes for the cycloaddition reaction (Scheme 2.26). In the presence of 10 mol% H8-BINOL-based phosphoric acid 6f, cinnamaldehydes 87, primary amines 7, and 1,3-dicarbonyls 88 underwent three-component cyclization reactions smoothly to give enantioenriched 4-aryl substituted 1,4-dihydropyridines 89 with high enantioselectivities of up to 98% ee [37]. The same group also developed the asymmetric synthesis of 3-amino 6-lactams 91 by phosphoric acid—catalyzed cyclization reactions involving azlactones 90 as both nucleophiles and electrophiles (Scheme 2.27) [38]. In addition to aromatic amines, substituted aryl ethylamines 92 participated well in such cyclization reactions to afford products 91a, which can be converted to benzo[a]quinolizidine derivatives 93 after being treated with trifluoroborane in high overall yields with excellent enan- tioselectivity, ranging from 90 to 97% ee [38].](https://figures.academia-assets.com/35916932/figure_102.jpg)



![2.2.13. Halocyclization The asymmetric halocyclization reaction represents one of the most important transformations [43] in the construction of enantioenriched heterocycles containing](https://figures.academia-assets.com/35916932/figure_106.jpg)
![Very recently, a phosphoric acid—catalyzed intermolecular bromoesterification reaction of carboxylic acids 108 and cyclohexene 109 carried out by Li et al. demon- strated for the first time the possibility of haloesterification between two substrates under organocatalyzed conditions, albeit in low yields and moderate enantioselectivities because of the competitive bromoesterification of the catalyst (Scheme 2.33) [47]. 2.2.14 Redox Reaction](https://figures.academia-assets.com/35916932/figure_107.jpg)


![More recently, a novel one-pot synthesis of enantioenriched polysubstituted -yclopenta[b]indoles 127 starting with the o-alkylation of aldehydes 125 was >stablished by Xu et al. [51]. In the presence of a primary-amine-derived thiourea T1 ind a carboxylic acid A1, the reaction between o,c-disubstituted aldehydes 125 and 3-indolylmethanol 124 proceeded smoothly to afford alkylation products, which inderwent two further consecutive Friedel-Crafts reactions catalyzed by a phosphoric icid PA1 to afford the desired polysubstituted cyclic products 127 with excellent stereoselectivities (Scheme 2.36).](https://figures.academia-assets.com/35916932/figure_110.jpg)
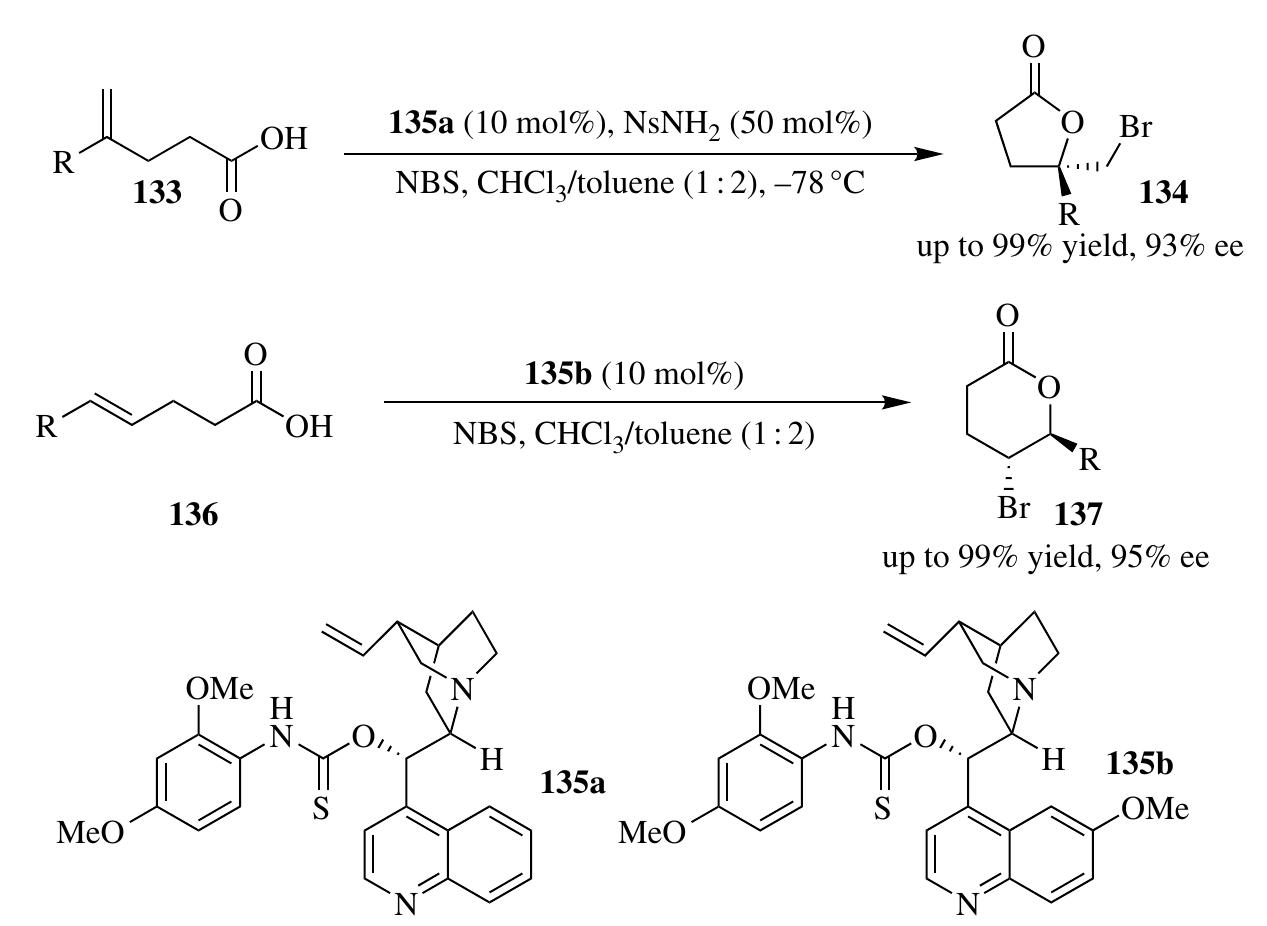
![2.3.1.1 Halolactonization The halolactonization reaction is an important synthetic method to construct halolactone from alkenoic acid substrates. The first example ot organocatalyzed asymmetric halolactonization reaction, reported by Wang et al. [44] was performed under phase-transfer catalysis with relatively low enantiocontrol The dramatic improvement in the enantioselectivities of halolactonization reactior was made by Whitehead et al. employing (DHQD),PHAL as the catalyst and DCDPH (1,3-dichloro-5,5-diphenylimidazolidine-2,4-dione) as the halogen source [53] Independently, Zhang et al. reported a chiral thiourea (130a)-catalyzed 1,4-bromolac- tonization of conjugated enynes 128 or 129, which afforded lactone heterocycles 131 or 132 bearing bromoallenes in high optical purity (Scheme 2.37) [54]. In another example of asymmetric organocatalytic bromolactonization of |,1-disubstituted alkenoic acids 133, reported by Zhou et al., Cinchona alkaloid— lerived aminothiocarbamate 135a was found to be an optimal sulfur Lewis base o activate NBS and control the enantioselectivity (Scheme 2.38) [55a]. Control -xperiments showed that both N-H and S of the thiocarbamate 135a were critical for ybtaining high enantiocontrol, indicating a Lewis base/hydrogen-bonding dual ictivation of NBS. Following similar strategy, the same group expanded the substrate sroup to 1,2-disubstituted alkenoic acids 136 with good stereocontrol by slight mod- fication of the catalyst (Scheme 2.38) [55b]. A high enantiocontrol iodolactonization via organocatalysis was achieved](https://figures.academia-assets.com/35916932/figure_111.jpg)



![Cinchona alkaloid thiourea (130d)-catalyzed cascade Michael—aldol reaction between 2-mercaptobenzaldehydes 151 and o,B-unsaturated oxazolidinones 152, leading to benzothiopyran derivatives 153 bearing three stereogenic centers with perfect levels of stereochemical control (Scheme 2.43a) [62a]. Following the same strategy, this group employed maleimides 154 instead of o,B-unsaturated oxazolidinones as Michael acceptors, obtaining succinimide-containing benzothio- pyrans 155 in good optical purity (Scheme 2.43b) [62b]. Another example of similar Michael—aldol reaction was reported by Dodda et al. in 2008, which involved readily available benzylidenemalonates 156 as Michael acceptors (Scheme 2.43c) [62c]. More recently, Dong et al. described a related approach by employing N-acyl pyrazoles 158 as the hydrogen-bond accepters of thiourea catalyst [62d]; both B-aryl- and f-alkyl-substituted o,f-unsaturated N-acylimides are found to be good paticipants to afford thiochromanes 159 with excellent stereoselectivities (Scheme 2.43d). Tae TUTE FE os seca scacounasdewawolo wes: pel Eis ea ers sew ace Bn we: EA deel ox Edel camxaercelias wesw](https://figures.academia-assets.com/35916932/figure_116.jpg)



![In 2012, Sun et al. established an enantioselective construction of spirocyclopentane bioxindoles from methyleneindolinones 54b and 3-substituted oxindoles 171 via a Michael-alkylation sequence (Scheme 2.47). The chiral 1,2-diaminocyclohexane—derived squaramide 172 was found to be the best choice of the catalyst, affording the desired products 173, containing three contiguous stereocenters in good stereoselectivities [66]. 2.3.1.5 Cyano-Involved Michael-Cyclization Reaction Cyanoolefins 174, o which both the electron-deficient double bond and the cyano group can be attacke: »y nucleophiles, are good substrates in tandem reactions to form heterocycles. Wan; >t al. reported that in the presence of bifunctional thiourea-tertiary amine 140b, th X,0-dicyanoolefins 174 and 2-naphthols 175 could undergo Michael addition to forn Friedel-Crafts intermediates, which then underwent intramolecular addition an ifforded corresponding naphthopyran derivatives 176 in high yields with moderat snantioselectivities (Scheme 2.48a) [67]. Another example of cyanoolefin-involve: andem reaction following a similar strategy was reported by Ding and Zhao, wh >mployed cyclohexane-1,2-dione 177 as a nucleophile (Scheme 2.48b) [68]. Zhao et al. also presented the asymmetric synthesis of multifunctionalizec](https://figures.academia-assets.com/35916932/figure_120.jpg)


![2.3.1.6 Michael-Hemiketalization (Hemiacetalization) Reaction Similar to cyanoolefins, «,B-unsaturated ketones bearing two electrophilic sites are also good substrates in a Michael-cyclization reaction. As an extension of work on cyanoolefin- involved naphthopyran synthesis [60], Wang et al. employed B,y-unsaturated o-keto esters 179 as electrophiles to react with 2-naphthols, which underwent Michael- hemiketalization to afford naphthopyran 187 with up 90% ee (Scheme 2.51a) [73]. In addition to 2-naphthols, coumarins 188 [74], trifluoroacetoacetates 164b [75], and 2-hydroxy-1,4-naphthoquinones 192 [76] participated well in this type of Michael- hemiketalization reaction in the presence of hydrogen-bonding thiourea catalysts (Scheme 2.51b to d). a 184 in good yields and enantioselectivities (Scheme 2.49b), while common enones ulways convert to normal Michael addition products. Both the catalyst and solvent were found to be critical to the oxa-Michael-tautomerization process [70]. In a sim- lar way, an enantioselective Mannich reaction/cyclization/tautomerization cascade sequence was developed that provided access to the 2-amino-4H-chromenes 184a ‘rom ot-amido sulfones 142b and malononitrile in good yields and enantioselectivi- ies (Scheme 2.49c) [71]. The presence of 10mol% rosin-derived thioureas 185a efficiently promoted a andam AAS nhanal acrnlinntinan mennnaa hatte an atrnnnnlafinn TWA anA 9D fremawAnnm](https://figures.academia-assets.com/35916932/figure_124.jpg)



![2.3.1.7 Michael-Henry Reaction Liu et al. and Xie et al. independently founc that tertiary amine-thioureas could stereoselectively promote the addition of diethy 0.-aminomalonate-derived azomethine ylides to nitroolefins, affording Michae adducts other than dipolar cycloaddition adducts as the major products. Using monofunctional chiral thioureas 140d instead of tertiary amine-thiourea catalysts Liu et al. successfully developed a three-component [3+2] dipolar cycloaddition o: benzaldehydes 3, diethyl a-aminomalonates 45a, and nitrostyrenes 165, resultins directly in the enantioenriched pyrrolidines 208 as the only products (Scheme 2.56 [81a]; while Xie et al. efficiently converted the Michael adducts 210 to pyrrolidine: 208 in high yield and maintained ee by the use of 30 equiv of 2,2,2-trifluoroethano as the additive (Scheme 2.56) [81b]. Under the nromotion of 270 mol% primary amine thioureas 140e and 20 mol% achira](https://figures.academia-assets.com/35916932/figure_128.jpg)


![The organocatalyzed asymmetric synthesis of bicylco[3.2.1]octan-8-ones 214 was described independently by Rueping et al. [84a] and Ding et al. [84b] respectively ‘Scheme 2.58). The bifunctional thioureas were found to be optimal catalysts to pro- mote the Michael—Henry cascade reaction of cyclohexa-1,2-dione 177 with the 3-nitrostyrenes 165, which afforded the bicycles desired, 214 in good yields and stereoselectivities.](https://figures.academia-assets.com/35916932/figure_131.jpg)
![Jia et al. reported a novel Michael/aza-Henry reaction of well-designed chalcones 217 and nitromethane 218 catalyzed by thiourea 130c (Scheme 2.60). Initiated by the Michael addition of nitromethane to an electron-deficient double bond, the resulting intermediate subsequently underwent an aza-Henry reaction to furnish multisubsti- tuted tetrahydroquinolines 219a with up to 20:1 dr and >99% ee [86a]. Later, the same group presented an alternative way to construct the same skeletons with different diastereoselectivities [86b], which employed nitrogen as the nucleophilic site to trigger the cascade process (Scheme 2.60). Very recently, a multifunctional](https://figures.academia-assets.com/35916932/figure_132.jpg)




![SCHEME 2.65 Asymmetric Michael—Michael reaction for the construction of cyclohexane BE ere gee caer More recently, an asymmetric domino Michael—Michael reaction between nitro- hex-4-enoates 240 and nitroolefins 165 was achieved successfully by Rajkumar et al., which allowed the efficient construction of cyclohexanes 241 with up to five stereocentres in high stereoselectivities (Scheme 2.65) [94a]. Using a similar strategy, Xu’s group furnished an efficient construction of the chiral tetracyclic core 244 of lycorine-type alkaloids in 63% yield over three steps (Scheme 2.65) [94b].](https://figures.academia-assets.com/35916932/figure_137.jpg)
![Nazarov reagents 238, which possess both a nucleophilic carbon and an electron- deficient C—C double bond, have been employed in the synthesis of enantioenriched spiro[4-cyclohexanone-1,3’-oxindoline] derivatives 239 by Wei and Gong. The Michael—Michael cascade reaction of 238 and methyleneindolinones 55 proceeded smoothly in the presence of the bifunctional urea catalyst 140h and 4-A molecular sieves, which afforded structurally diverse spirooxindole derivatives 239 with excel- lent enantioselectivities (Scheme 2.64) [93].](https://figures.academia-assets.com/35916932/figure_138.jpg)

![2.3.1.10 Sulfur Ylide—Involved Michael-Cyclization Reaction Lu et al. reveale that with the promotion of 10mol% simple achiral thiourea 258 and with 10 mol‘ DMAP as the cocatalyst, sulfur ylides 257 and nitroolefins could undergo an unprec edented cascade process to afford structurally diverse oxazolidin-2-ones 260 wit great levels of diastereocontrol (Scheme 2.67). The cycloaddition product 26 obtained from the addition of ethyl acrylate 261 to the reaction mixture verified th: the starting materials underwent Michael addition and oxygen alkylation to affor isoxazoline N-oxide 259 intermediates, which would then undergo epoxidatio1 deprotonation, ring opening, Hofmann rearrangement, and ring closing to yield th desired product [96]. Subsequently, a formal [4+ 1]/[3+2] cycloaddition cascade reaction with sulfur ylides and nitroolefin derivatives 263 was carried out in a similar manner by the same group. The isoxazoline N-oxide 259 intermediates underwent a series of intra- molecular reactions, affording fused heterocyclic structures 264 with excellen diastereoselectivities (Scheme 2.68) [97].](https://figures.academia-assets.com/35916932/figure_140.jpg)
![2.31.11 a-Isothiocyanato Imide-Involved Cascade Reaction The potential of Q@-isothiocyanato imide in organocatalyzed asymmetric cascade reaction was first demonstrated by Li et al. in 2008. In the presence of 5 mol% thiourea catalyst 140k, the a-isothiocyanato imide 265 and aldehydes 3 underwent aldol reaction and subsequent O—C cyclization to afford protected syn-B-hydroxy-o-amino acid deriv- atives 266 with high stereoselectivities (Scheme 2.69) [98].](https://figures.academia-assets.com/35916932/figure_141.jpg)
![Subsequently, the asymmetric aldol-cyclization reaction of o-isothiocyanato imides and a-keto esters 267 was reported independently by Jiang et al. [99a] and Vecchione et al. [99b], respectively, forming cyclic thiocarbamates 268 bearing quaternary stereogenic centers in high stereocontrol (Scheme 2.70). The substrate scope of this reaction extended to isatins 56 by Jiang et al. which provided access to enantioenriched phytoalexin analogs 269 with promising antipyretic activity (Scheme 2.70) [100].](https://figures.academia-assets.com/35916932/figure_142.jpg)

![Chen et al. reported that oxindole-type o-isothiocyanato imides 271 had high activities in the aldol-cyclization reaction with inactive simple ketones. With the oromotion of thiourea catalyst 1401, the cascade process of either aromatic or iliphatic ketones 206 with 3-isothiocyanato oxindoles 271 proceeded smoothly to ifford spirooxindoles 272 bearing a quaternary stereogenic center with perfect stereo- selectivity (Scheme 2.72) [102]. Cao et al. recently extended the electrophiles to electron-deficient carbon—](https://figures.academia-assets.com/35916932/figure_144.jpg)













![The enantioselective synthesis of taxol side chain and (—)-epi-cytoxazone were then accomplished with this type of dual catalytic asymmetric multicomponent reac- tion [120]. Starting with the recrystallized B-amino-c-hydroxy]l acid derivatives 318, a relatively concise synthesis of taxol side chain 319 was carried out in 18% overall yield with >99% ee, while (—)-epi-cytoxazone 320 was synthesized in four steps from corresponding intermediates with 32% overall yield (Scheme 2.87).](https://figures.academia-assets.com/35916932/figure_158.jpg)
![the imine substrates, which were attacked Dy Oxonium-ylide intermediates derived from alcohols, diazoacetates, and rhodium catalyst [117]. Later, the four-component process of this type of reaction, involving alcohols, amines, aldehydes, and diazoace- tates, was also investigated [118]. The phosphoric acid was found to be crucial to both the chemo- and stereoselectivity, due to its effect on imine formation and activation. Recently, Xu et al. describled a highly enantioselective Mannich-type three- component reaction of diazoacetophenones 315, alcohols 311, and imines 148 under the cocatalysis of Rh,(OAc), and phosphoric acid 5m (Scheme 2.86). In the presence of Rh,(OAc),, diazoacetophenones and alcohols formed oxonium ylides to serve as the enol equivalents of a-alkoxyl aryl ketones, which then underwent Mannich-type reaction with phosphoric acid—activated imines to produce enantioenriched B-amino- a-hydroxyl ketone products 316 [119].](https://figures.academia-assets.com/35916932/figure_159.jpg)
![Recently, the same group found that in addition to oxonium-ylides, the protic car- bamate ammonium ylides can be stereoselectively trapped by imines before a 1,2-proton shift [121]. Building on a similar activation model, a Rh,(OAc),/chiral Brgnsted acid—cocatalyzed three-component Mannich-type reaction of diazo com- pounds 317, carbamates 321, and imines 148 was developed successfully, providing a rapid construction of both syn- and anti-o.-substituted «,B-diamino acid derivatives 322 and 323 with high chemo- and stereoselectivity (Scheme 2.88). Although fruitful achievements have been made in ylide-involved stereoselective synthesis, the unstable and highly reactive zwitterionic intermediates have never been employed as nucleophilic participants in catalytic asymmetric transformations. Very recently, Qiu et al. have addressed this more challenging problem by catching this type of intermediate with activated imines [122]. Under an Rh,(OAc),/chiral Brgnsted acid cocatalysis system, zwitterionic intermediates generating from either inter- or intramolecular insertions of carbenoids into aromatic rings stereoselectively attacked the imines in the promotion of a phosphoric acid, efficiently affording poly- functionalized oxindole 325 and indole 327 derivatives with excellent diastereoselec- tivity and enantioselectivity (Scheme 2.89). "hs AnANTACALASEwe 1 DeAFATA Gli LERPA COSA BREAN GCATrhAaAmMmALA AMMAN Wha](https://figures.academia-assets.com/35916932/figure_160.jpg)

![2.4.2.1 Pd(0)/Bronsted Acid System In 2007, Mukherjee and List developed a highly enantioselective a-allylation of branched aldehydes by employing Pd/ Brgnsted acid cascade catalysis [125]. In the presence of phosphoric acid 5b, the condensation of branched aldehydes 36 and allyl amines 334 afforded allyl substi- tuted enamonium phosphate salt intermediates (X28), which were then converted to a cationic m-allyl-Pd complex and an enamine (X29) by the catalysis of Pd(0); next, nucleophilic attack of the m-allyl-Pd complex by the enamine occurred, affording allylated aldehydes 336 containing all-carbon quaternary stereogenic centers in high optical purity (Scheme 2.92). Besides, an enantioselective total synthesis of (+)-cuparene (338) was also accomplished using this strategy. Subsequently, efforts aimed at employing simple allylic alcohol 339 as the precursor of a 7-allyl-Pd com- plex intermediate in similar asymmetric transformations were made by the same group [126]; however, low enantioselectivities were obtained, possibly because of the poor E/Z ratios of enol intermediates derived from aldehydes (X30) [126]. The addition of 40mol% of benzhydryl amine is found crucial for the success of high enantiocontrol, which may convert the aldehyde into enamine with high E/Z selectivities (X31, Scheme 2.92).](https://figures.academia-assets.com/35916932/figure_163.jpg)


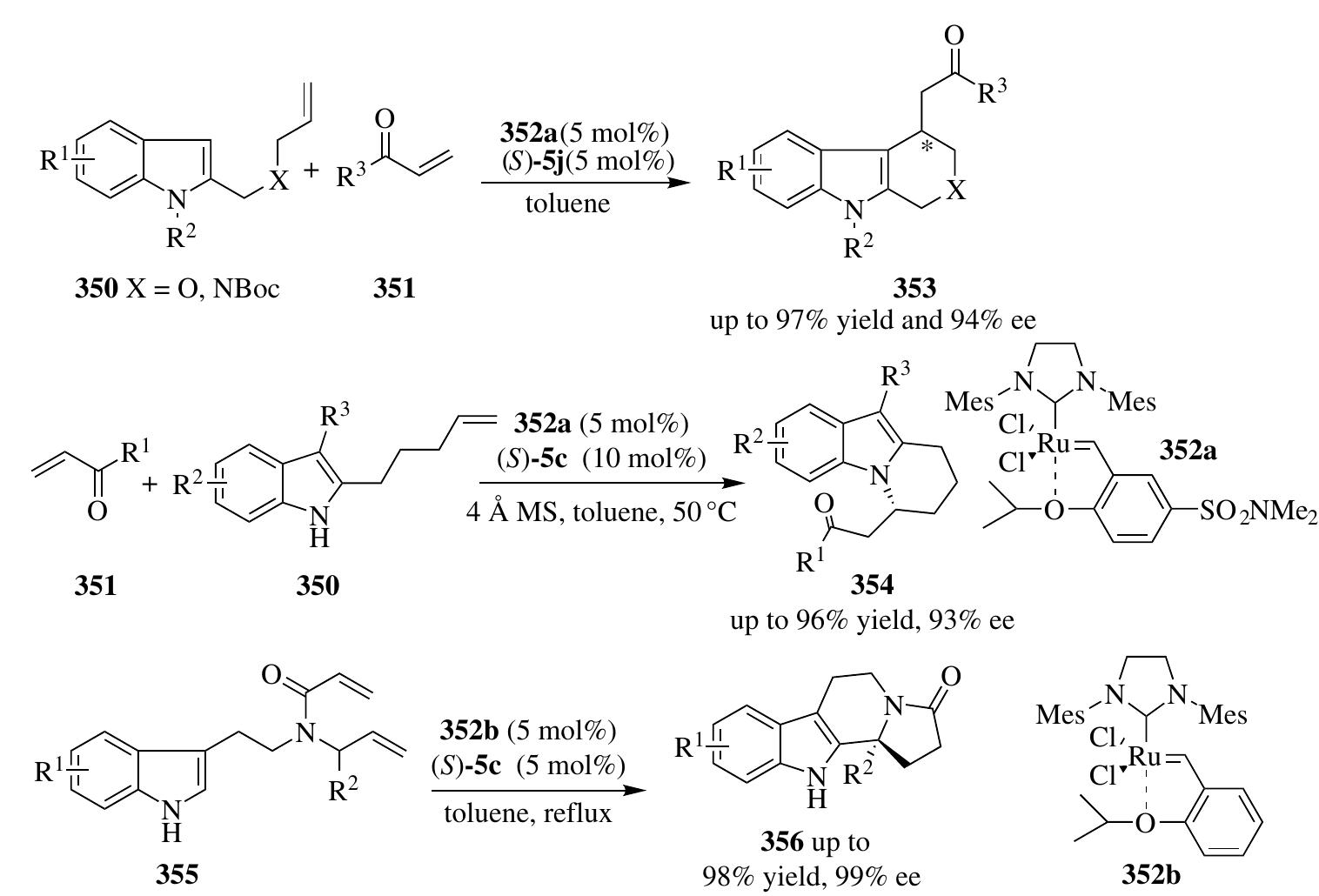

![cymene)I,], in giving racemic 358 was of great importance for stereocontrol. Inspired by this success, Chen et al. developed an efficient biomimetic asymmetri hydrogenation of benzoxazinones 361a by employing a catalytic amount of Hantzsc esters as the regenerable hydrogen source [130b]. The Hantzsch pyridine 363, whic was generated from the phosphoric acid—catalyzed asymmetric transfer hydrogen tion of benzoxazinones 361a with Hantzsch ester 37b, could undergo subsequer hydrogenation with H, in the presence of Ru complexes to regenerate Hantzsch este for the next catalytic cycle. The lower reaction rate of Ru-catalyzed direct hydroge nation of benzoxazinones is crucial for the success of enantiocontrol (Scheme 2.96 In a similar way, the same group has found that this [Ru(p-cymene)I,],/phosphori acid binary catalytic system enables 9,10-dihydrophenanthridine (DHPD) to act as new and easily regenerable NAD(P)H model in the asymmetric hydrogenation c 361b with wild substrate scope under mild conditions (Scheme 2.96) [130c]. 2.4.2.3 Au(I)/Brgnsted Acid System Han et al. developed an unprecedented protocol to synthesize tetrahydroquinolines 332 directly from 2-(2-propynyl)aniline derivatives 365 in one pot under relay catalysis of an achiral Au complex 368 and a chiral phosphoric acid 5j [131]. The Au'-catalyzed intramolecular hydroamination of 2-(2-propynyl)aniline provided the 1,4-dihydroquinolines 366, followed by isomeri- zation into imine-like 3,4-dihydroquinoliniums 367 with 5j. This active intermediate then underwent asymmetric transfer hydrogenation with Hantzsch ester to produce enantioenriched tetrahydroquinoline products (Scheme 2.97). Independently, Liu and Che reported the gold(1)/chiral Bronsted acid—catalyzed teinaes setarmmnlAnileas KemstnsemeR Se teeretax hHSASEMRHHAMA BARA Ot](https://figures.academia-assets.com/35916932/figure_168.jpg)










![steps, WE LOLdl syHUICSIs OF Hdatural PrlOQuct CalllplOuJeNCH OF Wds dUlleved,. An enantioselective formal synthesis of martinelline using a similar method was developed by Yoshitomi et al. (Scheme 3.6) [24]. Isolated from the roots of Martinella iquitosensis, martinelline 44 and martinellic acid 43 are effective nonpeptidic bradykinin receptor agonists [25]. Furthermore, the structure of these alkaloids possesses an unusually fused pyrrolidinotetrahydroquinoline core, which attracted more attention. Yoshitomi et al. developed a similar, yet more powerful strategy to construct the quinoline core 41 from simple precursors of unsaturated aldehyde 39 with o-aminobenzaldehyde derivative 38. The product desired, 41, was obtained in quantitative yield and 99% ee. Interestingly, a new route was developed for the total synthesis of (+)-ricciocarpin A (49) involving both an organocascade reaction and a Lewis acid—promoted cyclization sequence. Isolated from the liverwort Ricciocarpos natans, (+)-ricciocarpin A (49) is a furanosesquiterpene lactone that possesses potent molluscicidal activity against the water snail Biomphalaria glabrata, a vector of the parasitic disease schistosomiasis [26]. Based on the previous study of the reductive Michael-cyclization cascade reac- tion [27], Michrowska and List extended the study of this reaction for the asymmetric total synthesis of (+)-ricciocarpin A and discovered a more efficient reaction involving a samarium triisopropoxide—promoted epimerization—Evans—Tishchenko](https://figures.academia-assets.com/35916932/figure_180.jpg)


![SCHEME 3.12 Total synthesis of (+)-palitantin. 3.2.2.3 More Complex Cycle-Specific Cascade Catalysis Beyond the scope of the combination of enamine/dienamine with iminium-specific cascade catalysis, several other examples involved either the merger of enamine/dienamine with imin- ium catalysis into a single step or transition metal catalysis. Hong et al. carried out the total synthesis of (+)-palitantin based on an asymmetric organocatalytic cascade reaction (Scheme 3.12) [33]. (+)-Palitantin 70, isolated from Penicillium palitans and P. brefeldianum, is a polyketide metabolite which was found to display anti- fungal and antibiotic activity [34]. The new strategy developed for building the skel- eton is an L-proline-catalyzed self-condensation of o,B-unsaturated aldehyde 68. This cascade reaction might involve a dual activation: two molecules of the enal were activated by iminium ion and dienamine, respectively (Scheme 3.13). Then the nucleophilic dienamine attacked the electrophilic iminium species to generate the intermediate 71. Followed by an intramolecular Mannich type of reaction, the core structure 69 was obtained in 70% yield with 95% ee. After nine additional synthetic steps, total synthesis of target molecule 70 was achieved. By merging a transition metal-catalyzed reaction with an organocatalytic cascade BEBO A RAS SRR 2 nea ARBRE ABSA ASL wee lee | 6 BGs at A One eB OMS](https://figures.academia-assets.com/35916932/figure_183.jpg)




![CHEME 3.16 Total syntheses of (—)-angustureine, (+)-cuspareine, and (+)-galipinine. Strong Brgnsted acid catalysis has proven to be a powerful and reliable strategy for organic synthesis [37]. In the process of strong Brgnsted acid catalysis, the substrates are activated either by H-bond or protonation. As a result, the LUMO energy of the substrates is lowered and the nucleophilic addition becomes possible. Among the strong Br@nsted acids, chiral phosphoric acids are demonstrated to be the most reli- able catalysts and have been used in a wide range of organocatalytic reactions. One of these valuable transformations is the reduction of imines to produce optically active amines by Hantzsch ester hydride transfer [38]. Rueping et al. have applied Brgnsted acid catalysis successfully to the transfer hydrogenation of heterocycles such as quinolines and pyridines [39]. In these transformations, double-transfer hydrogenation cascade reaction occurred to afford the products desired with good yields and high enantioselectivities with excellent chemoselectivity. These powerful approaches have been used for the syntheses of a variety of nitrogen-containing heterocycles whose structural motifs are commonly found in many natural products. Several biologically active tetrahydroquinoline alkaloids, such as (—)-angustureine 88, (+)-cuspareine 89, and (+)-galipinine 90, were synthesized efficiently based on this double-transfer hydrogenation cascade reaction (Scheme 3.16). With regard to the mechanism (Scheme 3.17), quinoline 83 was activated by the formation of imin- ium ion in the presence of the Brgnsted acid catalyst 85. Then the first hydride transfer](https://figures.academia-assets.com/35916932/figure_188.jpg)



![The Amaryllidaceae alkaloids encompass a large number of natural products with potential pharmacological and/or biological activities [42]. Among them, lycorine type alkaloids represent an important subgroup of this family (Figure 3.2). Xu et al. developed an asymmetric bifunctional thiourea-catalyzed cascade reaction for the stereoselective construction of the tetracyclic core of lycorine-type alkaloids and the formal synthesis of a-lycorane (Scheme 3.20) [43]. As expected, the low reactivity of alkyl-substituted malonate 102 reacted with strong electron-donating nitroolefin 103 to construct C ring 104 successfully with an all-carbon quaternary carbon center. The key skeleton 106 was synthesized efficiently with a total yield of 63% in only three simple operations involving two consecutively cascade reactions. The tetracyclic core can be applied further in the formal synthesis of a-lycorane.](https://figures.academia-assets.com/35916932/figure_192.jpg)

![The rich gold chemistry has stirred up tremendous excitement in the synthetic ommunity and spurred the publication of many excellent reviews that cover the topic ither comprehensively [2] in its early stage or, today, increasingly in specific areas 1,3]. Although Kirsch reviewed elegantly heterocyclic formation via gold/platinum- atalyzed cascade reactions [3d], a general review of this topic is overdue, especially onsidering the ever-increasing number of transformations falling into the realm. This hapter is intended to fill the void. However, due to the sheer number of such reactions, he limited space, and the desire to keep it concise, we do not offer either a critical or . comprehensive treatise of this topic but, rather, present an organized collection of epresentative reactions that highlight the versatilities and synthetic potential of gold SLE VWISt PEULEDSSLD Ad All UVUULT. Homogeneous gold catalysis has experienced explosive development during the past decade or so. A large array of novel and versatile transformations have been developed based primarily on the following unique reactivities of gold (Scheme 4.1): (1) gold complexes are potent soft Lewis acids and can effectively activate alkynes and allenes toward attacks by a range of nucleophiles; and (2) the alkenylgold inter- mediates thus formed can be nucleophilic at either the proximal end (i.e., the Q-position) or the distal end (i.e., the B-position) of the C—C double bond, the latter leading to the formation of highly reactive gold carbenes. To date, gold catalysis has become an indispensable tool in the synthetic repertoire of organic chemists and has been used increasingly in the synthesis of complex molecules, including natural products [1].](https://figures.academia-assets.com/35916932/figure_194.jpg)
![Enynes are highly versatile substrates for gold catalysis. In gold-catalyzed enyne isomerization reactions, which are often cascade processes, the typical initial step is a nucleophilic attack of a gold-activated C—C triple bond by the tethered C—C dou- ble bond, leading to the homoallylic cation 1 (Scheme 4.2). Interestingly, this cation is mesomeric to the cyclopropyl gold carbene 2 as well as the corresponding carboca- tion 3, suggesting a phenomenon of “nonclassical” carbocation [2a]. These structures could undergo various productive transformations, and the overall process is often termed an enyne cycloisomerization. Notably, although other metals, such as Pt and Ga, could promote this process, homogeneous gold catalysts are the most effective [4]. Since Echavarren group’s seminal work in this area in 2004 [4], gold-catalyzed enyne isomerizations have exploded, and the variety and novelty of the transforma- tions have provided continuing inspiration for gold reactions involving other substrates. Readers interested in this research area should consult several authorita- tive reviews [3a,c,ag], but the relevant cascade reactions are highlighted selectively below. 4.2.1.1 Cascade Reactions of 1,6-Enynes The pioneering work of Hashmi et al. on homogeneous gold catalysis [5] offers an excellent example of the potent cou- pling between alkynes and alkenes. As shown in Scheme 4.3, the alkynylfuran 4 can be viewed as a functionalized 1,6-enyne. In the presence of AuCl, at ambient temper- ature, it undergoes cascade transformations, yielding the synthetically useful highly substituted phenol 5. The first step upon gold coordination is the reaction between the “en” from the furan ring and the “yne” moiety, leading to a reactive gold carbene intermediate, which can be described by the two isomeric forms 6 and 7. Structure 7 could undergo a series of ring reorganizations, eventually leading to the aromatized product.](https://figures.academia-assets.com/35916932/figure_195.jpg)


![to an elegant total syntheses of (+)-orientalol F by the same authors (Scheme 4.5) [7]. Mechanistically, an initial enyne cyclization of 14 promoted by gold could yield the cyclopropyl gold carbene 15, which is susceptible to a S.2 type of attack by the proximal carbonyl group. The oxocarbenium in the intermediate 16 would be attacked readily by the alkenylgold moiety, thereby yielding the tricyclic product 17. Notably, this [2+2+2] cycloaddition is highly diastereoselective, and the newly formed chiral centers in 17 are controlled by the chiral propargyl carbon center and the double-bond geometry. Similar strategies have been adopted for the synthesis of (—)-englerin A by Ma [8] and Echavarren [9]. With the C—C double bond fully substituted at the end proximal to the alkyne, the snyne moiety tends to cyclize in a 6-exo-dig manner, forming a relatively stable lertiary carbocation-containing intermediate (i.e., 20, Scheme 4.6), which can be trapped by nucleophiles in a cascade process. This is exemplified in elegant work by Sethofer et al. [10], where the cascade leads to the formation of three fused rings with high enantiomeric access when a chiral gold complex is used.](https://figures.academia-assets.com/35916932/figure_198.jpg)

![4.2.1.3 Cascade Reactions of 1,4-Enynes With the 1-bonds separated by only one methylene group, the typical cyclopropyl gold carbene intermediates could not be accessed. However, Buzas and Gagosz [18] reported that 1,4-enynyl acetates such as 32 could still lead to the formation of bicyclo[3.1.0]hexane products such as 33 (Scheme 4.9), reminiscent of the results with 1,5-enynes (e.g., Scheme 4.7). Mechanistically, this reaction begins with a gold-catalyzed 3,3-rearrangement of the propargyl ester moiety, reported previously and recently reviewed by Zhang et al. and Shen [3a,h]. The allenene intermediate 34 is activated further by the same gold catalyst, promoting double cycliza- tion to form the cyclopropyl gold carbene 35. Interestingly, this gold carbene resembles intermediates 23 and 29 and undergoes a 1 ,2-C—H insertion to afford the product observed.](https://figures.academia-assets.com/35916932/figure_200.jpg)

![racemization and affords chiral the cyclopentenone product with limited ee erosion. In an earlier study by Shi et al. (Scheme 4.10), 1,4-enynes with a regioisomeric acyl- oxy group (e.g., 36) undergo gold-catalyzed Rautenstrauch rearrangements, yielding cyclopentenone products (e.g., 37) [19]. Notably, this reaction is dramatically different from the one by Buzas and Gagosz above and, moreover, shows intriguingly high stereo- selectivity. DFT calculations by Faza et al. [20] provide a mechanistic rationale for the high stereoselectivity: Alkenylgold intermediate 38 is formed initially via 5-exo-dig cyclization by the pivaloxy group and subsequently undergoes C—O bond fragmentation to form pentadienyl cation 39. Interestingly, the helix chirality of 39 could keep the memory of the original center chirality in 38. The fast conrotatory cyclization prevents racemization and affords chiral the cyclopentenone product with limited ee erosion. 4.2.1.4 Cascade Reactions of 1,3-Enynes Zhang and Wang reported yet anothe isomeric enynyl ester that proceeds through a different reaction pathway [21]. A shown in Scheme 4.11, propargyl acetate 40 with an 1,3-enyne moiety is the sub strate and can undergo a gold-catalyzed 3,3-rearrangement. The carboxyallene thu formed (i.e., 43) could be activated further by the same gold catalyst, to form th oxocarbenium intermediate 44, which is also a pentadienyl cation due to the attache C—C double bond and can undergo a Nazorov type of reaction. The cyclic gold car pene intermediate 45 thus formed proceeds through sequential 1,2-C—H insertiot and hydrolysis, affording a cyclopentenone product. Interestingly, Lemiére et al [22], showed later that the gold carbene 45 can be diverted exclusively to cyclo propanate a tethered alkene when R is but-3-en-1-yl, yielding the tetracyclic produc 42 in 98% yield and with excellent diastereoselectivity. In 2NNR Tin and Yamamoto [932] renorted a neefil cascade reaction of ketone](https://figures.academia-assets.com/35916932/figure_202.jpg)

![SCHEME 4.13 Gold-catalyzed cycloisomerization of N-(pent-2-en-4-ynyl) amides. In their continued exploration of gold-catalyzed phenol synthesis, Hashmi et al. [26] reported an interesting in situ construction of the requisite furan moiety for subsequent phenol formation, and both processes are catalyzed by IPrAuCl/AgSbF, (Scheme 4.14). This cascade reaction highlights the versatility of gold catalysis and the power of rationale design in achieving high synthetic efficiency. Notably, the phenol OH group is regioisomeric to that of 5, which is controlled largely by the location of the methyl group.](https://figures.academia-assets.com/35916932/figure_205.jpg)

![SCHEME 4.16 Gold-catalyzed tandem transformation of 1-en-8-yn-4-ols. ALLO TIMIOCUIA OF Livi — Tl). Barluenga et al. [29] reported a tandem cyclization/Prins type of reaction of 1-en- 8-yn-4-ols. As shown in Scheme 4.16, hydroalkoxylation of the C—C triple bond is the fist step. The enol intermediate 62 thus formed could be protonated to form the oxocarbenium species 63. With the C—C double bond positioned ideally for a Prins type of cyclization, such a process indeed ensues, resulting in the formation of the synthetically useful bicyclic bridged ether 61 in an excellent yield.](https://figures.academia-assets.com/35916932/figure_207.jpg)
![Propargyl carboxylate is a versatile substrate for gold catalysis. In the presence of a gold catalyst, it would initially undergo either a 1,2-acyloxy migration (Scheme 4.17) or a 3,3-rearrangement. The former process leads to an alkenylgold carbene intermediate (e.g., 67), and the latter to a carboxyallene, as shown in Schemes 4.9, 4.11, and 4.15. The reaction outcome is drastically different depend- ing on the reaction pathways, the selectivity of which is controlled primarily by the substitution patterns on the propargyl moiety. In general, sterically and/or electronically unbiased substrates undergo reactions via an initial 3,3-rearrangement (e.g., Schemes 4.9, 4.11, and 4.15), while electronically [30] and/or sterically biased substrates prefer 1,2-acyl migrations. Scheme 4.17 shows an example reported by Shapiro and Toste [31]. The propargyl benzoate 64 has a gem-dimethy] group at the propargyl position but none at the alkyne terminus. As a result of this drastic steric difference, it undergoes 1,2-benzoxy migration selectively to form the gold carbene 67, which is a Fischer-type carbene and electrophilic. Its stepwise annulation with the enimine 65 affords the azepine product 66 in a synthetically useful yield.](https://figures.academia-assets.com/35916932/figure_208.jpg)


![corroborating the intermediacy of 83. O the scatrold of trondosins A and b. In 2007, Luo and Schreiber [35] reported a rapid synthesis of complex &-pyrones ria Au-catalyzed coupling reactions (Scheme 4.20). Importantly, o-pyrones are core ements found in many biologically active compounds. The readily accessible ropargyl propiolate 80 first undergoes an Au-catalyzed 3,3-rearrangement [3ah] see Schemes 4.9, 4.11, and 4.15), thereby generating the enyne allene 82 in situ in a eversible manner. Interestingly, the propiolate alkyne in 82 is activated selectively wer its allene moiety, thereby promoting a 6-endo-dig cyclization where the allene cts as a nucleophile. The oxocarbenium 83 thus formed can be trapped by a variety yf nucleophiles, including 5-iodoindole, to afford polysubstituted o:-pyrones (e.g., 31). Notably, starting from enantioenriched 80 (98% ee), racemic 81 is obtained, corroborating the intermediacy of 83.](https://figures.academia-assets.com/35916932/figure_211.jpg)
![chemistry. As shown in Scheme 4.21, tandem gold-catalyzed 3,3-rearrangement of the propargylic acetate 84 and further Au activation of the allene moiety thus formed gen- erates the oxocarbenium 88, which can readily be hydrolyzed into the intermediate 89. By using Selectfluor as a uniquely effective oxidant, the Au(I) center in 89 can be oxidized into Au(III) in the intermediate 90. This oxidation is probably facilitated by the anionic |-acylalkenyl ligand, which makes the gold center relatively electron-rich. Transmetallation from phenylboronic acid to 90 then provides the Au(III) complex 91, which undergoes reductive elimination to yield the cross-coupling product 85. This oxidative cross-coupling reaction opens up a novel area for Au catalysis and bridges contemporary Au catalysis based on alkyne/allene substrates and the well-established late transition metal-catalyzed cross-coupling reactions. The enone dimer side prod- uct 86 is due to a competing transmetallation by the Au(I) intermediate 89 [37], and the enone 87 is due to competitive protodeauration of 89. Very recently, Cai et al. [38] devised a gold-catalyzed [3 + 2] cycloaddition/hydro- lytic Michael addition/retro-aldol reaction cascade using propargylic ester substrates tethered to cyclohexadienones. Starting from the propargylic esters 92, as shown in Scheme 4.22, an Au-catalyzed 3,3-rearrangement generates the allene species 94, which is activated by the same gold catalyst. Nucleophilic attack of the allene moiety by the tether carboxylate and concomitant allylic elimination of the gold catalyst yields a 1,3-dipole 95, which then undergoes [3+2] cycloaddition with the enone](https://figures.academia-assets.com/35916932/figure_212.jpg)
![SCHEME 4.22 Gold-catalyzed [3+2] cycloaddition/hydrolytic Michael addition/retro- aldol cascade.](https://figures.academia-assets.com/35916932/figure_213.jpg)
![Another example of cascade reactions involving an initial gold-catalyzed 3,3-rearrangement of propargyl ester was reported recently by Lebceuf et al. [40]. As shown in Scheme 4.23, the allene intermediate 104, formed upon the initial step, indergoes 5-exo-dig cyclization to generate the oxonium 105, which then undergoes irst an acyl group rotation and then a 1,5-sigmatropic acy] shift. This unprecedented icyl shift is supported by DFT computations, and the final product, the dienone 103, s formed in almost a quantitative yield.](https://figures.academia-assets.com/35916932/figure_214.jpg)


![A surprising formal [4+2] cycloaddition on an s-trans-heterodiene framework was reported by Teng et al. (Scheme 4.26) [43]. In this interesting work, an initial gold-promoted cyclization of alkynyl benzaldehyde 116 leads to either oxocarbe- nium 118 or its mesomeric isomer, gold carbene 119. The reaction could proceed via both resonance extremes. While direct [4+2] cycloaddition between 118 and ethyl vinyl ether is less likely, concerted [3+2] cycloaddition with the gold carbene 119 would afford the tricyclic gold carbene 120, which upon 1,2-alkyl migration would offer access to the desired oxocarbenium intermediate 121. The collapse of 121 pro- vides the highly strained anti-Bredt oxacycle 117 in an excellent yield. In their continued study using ortho-tethered nitrone as the internal oxidant [44], Yeom et al. [45] reported the coupling of this intramolecular redox process with a pinacol/Mannich—Michael cascade. As shown in Scheme 4.27, an initial attack at the gold-activated C—C triple bond by the tethered nitrone generates the alkenylgold 124, which can undergo heterofragmenation of the N—O bond, facilitated by the electron- donating gold, and generate a reactive &-oxo gold carbene intermediate (i.e., 125). This type of alkyne oxidation process, originally reported by Shapiro and Toste [46] and Li and Zhang [47] independently, can be considered as a variant of the general reactivity of gold chemistry leading to the gold carbene formation discussed in Scheme 4.1 and have tremendous synthetic potential. With a tertiary alcohol next to the electrophilic carbene center, a pinacol-type rearrangement occurs readily, leading to the B-diketone 126. Notably, this type of pinacol rearrangement was previously realized by Li and Zhang [47]. Subsequent tandem Mannich reaction and Michael addition yield a com- plex tetracycle (i.e., 123) in a good yield and with good diastereoselectivity. By combining an initial gold-catalyzed hydroaminative indole formation with repetitive hydroarylations, Hirano et al. [48] reported the gold-catalyzed direct ee ee ee ee ee Dey ee Le es Ce: LS ee Se ey ey a! ee i %, ect Sn ME ok Mee pamemnercer rey](https://figures.academia-assets.com/35916932/figure_217.jpg)

![nucelophile to attack the ideally positioned gold-activated o-alkyne in a typically 5-endo-dig manner [50], forming an indole core with dinitrogen bonded to the indole nitrogen. Ready expulsion of dinitrogen, assisted by the gold moiety, generates an o- imino gold carbene intermediate (i.e., 133), which is highly electron deficient at the carbene center. Its reaction with allyl alcohol, followed by a Claisen rearrangement that is probably gold-promoted, provides a facile excess to indoxy] 131 in excellent](https://figures.academia-assets.com/35916932/figure_219.jpg)


![SCHEME 4.32 Au(1)-catalyzed formation of N-acyl iminium ion and the subsequent Pictet-Spengler-type reaction. In 2007, Yang et al. [54] reported gold-catalyzed rapid and efficient access to multiring heterocyclic compounds. As shown in Scheme 4.32, the initial gold-cata- lyzed cyclization of the alkynyl carboxylic acid 145 yields the 5-lactone 148, which is reactive toward nucleophilic ring opening, due to its enol ester nature, and readily undergoes aminolysis with the primary amine 146 to give the ketoamide 149.](https://figures.academia-assets.com/35916932/figure_222.jpg)
![SCHEME 4.31 Gold-catalyzed tetrahydropyran synthesis from homopropargylic ethers. In 2006, Jung and Floreancig [53] reported an efficient synthesis of tetrahydropyran from homopropargylic ethers. As shown in Scheme 4.31, the cationic gold complex, Ph,PAUNTE,, first catalyzes facile hydration of the alkyne 141 to form the methyl ketone 143. Probably facilitated by the acidic gold catalyst, elimination of methanol from 143 occurs, and the resulting enone 144 then undergoes an intramolecular Michael addition, probably promoted by the acidic gold complex, to produce synthetically use- ful tetrahydropyran 142 in 97% yield and with exclusive cis selectivity.a](https://figures.academia-assets.com/35916932/figure_223.jpg)

![SCHEME 4.34 Au-containing all-carbon 1,3-dipoles: generation and [3 +2] cycloaddition. As shown in Scheme 4.34, the dipole formation is accomplished via a gold-pro- moted, unprecedented migration-fragmentation sequence of the propargyl ketal 159. The prior intermediate 162 could undergo 5-endo-trig cyclization, which, how- ever, is disfavored according to the Baldwin’s rule [58]. Consequently, the dihydrofu- ran product 160 is formed in <5% yield. Instead, the 1,3-dipole 163 would probably be formed from 162 via the cleavage of acetone. Its rapid trapping by anisaldehyde would form the intermediate 164, which has a significant mesomeric isomer 165. Importantly, this isomer can undergo a favored and probably facile 5-exo-dig cycli- zation to afford the desired [3 +2] cycloadduct 161 with high diastereoselectivity.](https://figures.academia-assets.com/35916932/figure_225.jpg)
![SCHEME 4.35 Gold-catalyzed cascade construction of pyrrolo[1,2-a]quinolin-1-(2H)-ones.](https://figures.academia-assets.com/35916932/figure_226.jpg)
![SCHEME 4.36 Gold-catalyzed intramolecular oxygen transfer via a [4+2] pathway. Recently, Liu et al. [61] reported the efficient synthesis of highly substituted cyclo- pentenyl ketones. As shown in Scheme 4.36, the reaction apparently proceeds through a formal metathesis between the C—C triple bond and the tethered ketone carbony] group, which is rather intriguing. Isotopic experiments and theoretical calculations satisfactorily support the cascade mechanism outlined in Scheme 4.36: An initial gold-promoted 5-endo cyclization forms the five-membered oxocarbenium 174, which undergoes an intramolecular [4+2] cycloaddition with the tethered carbony] group to form the bridged intermediate 175. Subsequent consecutive ring openings would lead to the enone product 173. Notably, this is the first example of [4+2] cyclo- addition between an O-alkenyl oxonium intermediate and a carbonyl group.](https://figures.academia-assets.com/35916932/figure_227.jpg)

![in 90% yield upon treating enallenyl TMS ether 183 with an Au(IID) complex [i.e., dichloro-2-picolinoatogold(II)]. Mechanistically, selective activation of the enolic dou- ble bond of the allenyl ether moiety by the Au(II) complex would give the oxocarbenium 185, which is in resonance with the 1,3-dipole 186. An intramolecular 1,3-dipolar cyclo- addition between the dipole moiety and the C—C double bond in 186 would generate the Au carbenoid 187, which is strained due to the bicyclo[3.1.0Jhexane structure. Facile fragmentation of the cyclopropane ring would probably ensue, facilitated by the release of ring strain as well as the TMSO group. The aurated intermediate 188 thus formed would then proceed through sequential desilylation by water, generation of acid, and protodeauration to the isolated bicyclic product. It is important to note that this reaction is stereospecific in relation to the double-bond geometry, and the product contains an all- carbon quaternary center with two differentiated carbonyl groups. This feature might lend this chemistry to applications in complex molecule synthesis. In 2009, a novel dual metal catalysis involving Au and Pd was reported by Shi et al. [64]. In this catalyzed catalysis, a soft Lewis acidic Au complex transforms a substrate tm 7 .- ya A AN](https://figures.academia-assets.com/35916932/figure_229.jpg)


![Early in 2011, Zhang et al. [66] reported the first oxidative cross-coupling reaction between an aryl C—H and an in situ—generated alkylgold by employing an Au(I)/ Au(III) catalysis. As shown in Scheme 4.41, the reaction constitutes a formal [3+2] annulation between the tethered vinyl group and the aniline moiety. Mechanistically, the reaction begins with an intramolecular aminoauration of the vinyl group in a highly diastereoselective anti-addition manner [67]. The intermediate thus formed (i.e., 204) can be isolated and characterized in a stoichio- metric reaction. Subsequent Selectfluor oxidation of 204 into the Au(IID) complex 205 should be followed by a facile electrophilic aromatic auration, thereby gener- ating the six-membered Au(IID intermediate 206. This Au(III) complex could then undergo a concerted reductive elimination with retention of the stereochemistry to afford the tricyclic indoline product 203. Notably, this mechanism is consistent with deuterium labeling experiments. In the presence of gold complexes, cyclopropenes can be converted into syntheti- cally versatile alkenyl gold carbene intermediates, which can be of high synthetic utility [3a,b]. In 2011, Hadfield and Lee [68] reported a cascade process utilizing this inter- mediate, where a series of functionalized trienes were formed via facile Au(I)-catalyzed reactions between cyclopropenes and furans. Scheme 4.42 shows an example and the proposed mechanism: an initial Au-activated ring opening of the cyclopropene 207 generates the gold carbene 209, which then reacts with furan to form the](https://figures.academia-assets.com/35916932/figure_232.jpg)

![SCHEME 5.1 Ru-catalyzed cascade RCM/hydrogenation and RCEM/hydrogenation. Recently, Yoshida et al. reported a cascade RCEM/RCM with Grubbs’ second- generation catalyst 2 (7.5 to 15 mol%), which allowed consecutive construction of two rings from tetraenynes 4 (Scheme 5.2) [9]. After dehydration with p-TsOH (15 mol%) at room temperature, the resulting diol 5 could be converted to biaryl compounds.](https://figures.academia-assets.com/35916932/figure_234.jpg)

![The cascade process reported by Varela et al. [14] involved a Ru-catalyzed addition of alkenes 15 to 1,6-diynes 16 and a thermal 67 electrocyclization (Scheme 5.7). A reaction producing polycyclic cyclohexadienes 17 is performed under a catalytic mixture of 10% [Cp*Ru(CH,CN),]PF, and 10% Et,NCI at 80°C. The Ru-catalyzed cascade isomerization/Claisen rearrangement reaction was demonstrated to be very efficient for the transformation of 1,7-dienes 18 into y,](https://figures.academia-assets.com/35916932/figure_236.jpg)
![In the presence of diazo compounds 9, enynes 10 containing a fluorinated amino acid moiety could be transformed into fluorinated alkenyl bicyclo[4.1.0]heptane amino acid derivatives 11 using Cp*(Cl)Ru(COD) as the precatalyst (Scheme 5.5) [12]. In this process, the in situ-generated catalyst from ruthenium complex and diazo compound completely inhibits RCM of enyne to the profit of cascade alkenyl- ation/cyclopropanation. The Cp*(Cl)Ru moiety in ruthenacyclobutane is believed to favor reductive elimination versus expected alkene metathesis.](https://figures.academia-assets.com/35916932/figure_237.jpg)
![SCHEME 5.9 Ru-catalyzed cascade Meyer—Schuster rearrangement/aldol condensation. Lately, a Ru-catalyzed cascade reaction involving Meyer—Schuster rearrangement of propargylic alcohols and an aldol condensation was discovered (Scheme 5.9) [16]. The 16-electron allyl ruthenium(II) complex [Ru(n*-2-C,H ,Me)(CO)(dppf)][SbF,] was used as a catalyst. Using this method, a large variety of conjugated dienones 20 and diene—diones 21 were synthesized from terminal propargylic alcohols and enoliz- able ketones (Scheme 5.9) or B-dicarbonyl compounds (Scheme 5.10), respectively.](https://figures.academia-assets.com/35916932/figure_238.jpg)




![Catalytic asymmetric Michael addition is an important reaction for creating carbon-carbon bonds with enantioselectivity. This reaction can be combined with other catalytic transformations to build up complex organic structures. A successful example is the enantioselective cascade Michael addition/H,-hydrogenation catalyzed by ruthenium hydride borohydride complexes containing B-aminophosphine ligands 26 (Scheme 5.13) [19]. This approach has been extended to pentenones, heptenones, and nitrostyrene Michael acceptors and malonitrile Michael donors.](https://figures.academia-assets.com/35916932/figure_243.jpg)


![Guo et al. [24] reported iron-catalyzed tandem oxidative coupling and annulation of phenols and f-keto esters, furnishing polysubstituted benzofurans 38 (Scheme 5.20). The combination of FeCl,-6H,O and (t-BuO), offers an efficient catalytic oxidative system. The iron catalyst demonstrates dichotomous catalytic behavior in this transformation, which is a transition metal catalyst in the oxidative coupling step and a Lewis acid in the condensation.](https://figures.academia-assets.com/35916932/figure_246.jpg)
![SCHEME 5.19 Fe(III)-catalyzed cascade cyclization/halogenation of alkynyl diethyl] acetals.](https://figures.academia-assets.com/35916932/figure_247.jpg)
![SCHEME 5.21 Fe(ID)-catalyzed three-component reaction of alkenes, aldehydes, and hydroperoxides. More recently, Qin et al. reported an iron-catalyzed transformation of benzyl hydrocarbons into corresponding amides through C—H and C—C bond cleavage under mild reaction conditions (Scheme 5.23) [26]. The cascade process involves an oxidative rearrangement. As shown in Scheme 5.24, diphenylmethane undergoes iron-assisted single-electron-transfer (SET) oxidation with DDQ to produce the corresponding diphenylmethyl radical A, which could additionally be oxidized to the diphenylmethy] cation B. Then substitution reaction of B with an azide anion gener- ates C, which is oxidized to diphenylmethy] azide cation D by the iron-assisted DDQ](https://figures.academia-assets.com/35916932/figure_248.jpg)


![Two different metal-catalyzed cascade processes have also been reported. A recent example is the FeCl,/PdCl,-cocatalyzed coupling cyclization of 2,3-allenoates 41 with allylic bromides 42 (Scheme 5.25) [27]. This protocol provides very concise access to B-allylic-substituted butenolides 43. The key point for this catalyst relaying process is the in situ generation of sp’ carbon iron species A and its transmetallation with PdCl, Another successful catalyst relaying process was discovered in iron—copper cooperative catalysis in the exchange reaction between cyclopentylmagnesium bromide and terminal alkenes and the alkylmagnesiation of alkynes [28]. The Grignard exchange between terminal alkenes (RCH=CH,) and cyclopentylmagne- sium bromide was catalyzed by FeCl, and CuBr in combination with PBu, to give RCH,CH,MgBr, which could be capiimed by an electrophile (Scheme 5. 26).](https://figures.academia-assets.com/35916932/figure_251.jpg)









![The combination of reactions of rhodium carbenoids with polyether—macrocycl synthesis offered interesting procedures for the synthesis of this important clas of compounds. One elegant example is the Rh-catalyzed four-component reactio: of two o-diazo-B-keto esters and two cyclic ethers, such as tetrahydrofuran o 1,4-dioxane, to yield functionalized 16- to 18-membered macrocycles 6: (Scheme 5.44) [42]. The process involves the generation of electrophilic rhodiun carbenoid A, the addition of cyclic ether to this intermediate, as well as the formatio: and dimerization of the oxonium ylide intermediate B. Another example is th Rh-catalyzed macrocyclization of oxetanes with o-diazocarbonyls (Scheme 5.45 [43]. In this case, three oxetanes and one rhodium carbenoid intermediate condens: in a one-step process. It is noteworthy that these macrocyclizations could procee under high-concentration conditions (1M). Pliaxtwins elas: Aati atest @¢603oeeclew: evan wes ToaslhewelT 9 4 bdcoerlae 3 xml:](https://figures.academia-assets.com/35916932/figure_261.jpg)





![SCHEME 5.52 Rh-catalyzed 1,3-acyloxy migration/[5 + 1] cycloaddition reaction. Rhodium catalysts could also promote the addition of arylborates and arylboronic acids to C=C and CXO bonds. The Rh-catalyzed addition—cyclization of sodium tetraarylborates 80 to 3-(2-alkynylethyl)-2-cycloalken-1-ones 79, furnishing spiro- carbocycles 81 was reported by Shintani et al. (Scheme 5.53) [51]. These tetraaryl- borates function as surrogates of |,2-dimetalloarenes, forming two new C—C bonds sequentially through the cascade process.](https://figures.academia-assets.com/35916932/figure_267.jpg)
![Another example is the Rh-catalyzed cascade approach to phthalides 84 from commercially available phthalaldehyde 82 and arylboronic acids 83 (Scheme 5.54) [52]. The first step of this transformation is the aryl addition of phthalaldehyde 82 with arylboronic acids 83, which leads to the formation of 2-(hydroxy(phenyl) methyl)benzaldehyde A. The second step probably involves an aldehydic C—H activation/C—O coupling.](https://figures.academia-assets.com/35916932/figure_268.jpg)

![The cascade process combining an Rh-catalyzed alkyne arylation and a Pd-catalyzed C—N coupling in a single vessel was discovered by Panteleev et al. (Scheme 5.55) [53]. This is a rare example of the catalyst relaying system, in which two transition metal complexes with different phosphine ligands capable of dissociation function along a desired pathway, even when other reaction pathways are available.](https://figures.academia-assets.com/35916932/figure_270.jpg)





![SCHEME 5.62 Four-component reactions of sulfonyl azides with terminal alkynes, alcohols, and nitroalkenes or Baylis—Hillman adducts. Recently, two four-component reactions of sulfonyl azides, terminal alkynes, nucleophilic coupling partners, and electrophilic acceptors were developed. One ele- gant example is the cascade synthesis of y-nitroimidates 95 (Scheme 5.62), which was reported independently by Wang’s [62] and Chang’s [63] groups. This process involves nucleophilic addition of alcohol to the in situ-generated ketenimine and Michael addition of the resulting carbon anion to the electron-deficient nitroalkene. Another example is the regioselective synthesis of 4-(alkoxycarbony1)-pent-4-enimi- dates 97 from Baylis—Hillman adducts 96 (Scheme 5.62) [64].](https://figures.academia-assets.com/35916932/figure_276.jpg)
![In addition to the heteroatom nucleophiles, carbon nuleophiles such as pyrroles were found by Cho and Chang [65] to be able to take part in the three-component reaction with sulfonyl azides and terminal alkynes to give the dual-functionalized pyrroles 98 (Scheme 5.63). The reaction depended on the presence of the N—H bond. When N-substituted pyrroles were used, reactions were sluggish and gave poor yields. Moreover, either indole or N-methyl indole did not participate in the reaction. Recently, Wang et al. [66] found that 2-methylindoles was workable toward the ketenimine (Scheme 5.64). In this case, the alkyl group at the 2-position of indole ring increased the electron density of the 3-carbon and made the copper-catalyzed three-component reaction possible. The reaction was sensitive to oxygen. When the reaction was exposed to air, the oxidized products 99 were isolated. The cascade pro- cess is believed to involve a CuAAC, a nucleophilic addition, and a copper-catalyzed C—H bond activation—oxidation. three-component reaction possible. The reaction was sensitive to oxygen. When the](https://figures.academia-assets.com/35916932/figure_277.jpg)
![The cascade reactions involving cycloaddition of ketenimine were also investi- rated. For example, the copper-catalyzed reaction of imines with terminal alkynes and sulfonyl azides undergoes a [2+2] cycloaddition of imines to the in situ-generated cetenimine, producing N-sulfonylazetidin-2-imines 100 with both high regioselectiv- ty and stereoselectivity in favor of the trans configuration except for imines derived rom ethyl glyoxylate (R*=CO,Et) (Scheme 5.65) [67]. Changing the imine substrates o o,B-unsaturated imines led to the [4+2] cycloaddition products 101 [68].](https://figures.academia-assets.com/35916932/figure_278.jpg)


![The cascade three-component coupling of aldehydes, terminal alkynes, and amines is one of the most convenient methods for the synthesis of propargylic amines with water as the only theoretical by-product. A recent example is the three- component coupling of aldehydes, alkynes, and carbamates with copper(ID triflate as catalyst, which furnished a diverse range of propargylcarbamates 103 in moderate to high yields, and no other cocatalyst or ligand is required in this transformation (Scheme 5.68) [71]. eee See Ae be ee Bhuvaneswari et al. [75] reported a copper-catalyzed three-component coupling of arynes, terminal alkynes, and activated alkenes, providing 1-alkyl-2-alkynylben- zenes 108 (Scheme 5.72). 2-(Trimethylsilyl)-3,4-dimethylpheny] triflates 107 were used as benzene precursors. Reaction of terminal alkynes with Cu(I) species in the presence of CsF (or K,CO,) gives copper acetylides A. Alkynylcupration of the in situ-generated arynes B with cuprous acetylides A affords arylcuprous intermediates](https://figures.academia-assets.com/35916932/figure_281.jpg)









![SCHEME 5.81 Cu(II)-catalyzed synthesis of aryl nitriles using ammonium iodide and DMF as a cyanide source. More recently, Kim et al. combined the copper-catalyzed C—H and C—B bon ictivation with the copper-catalyzed generation of cyanide anion in one pot t levelop an efficient synthesis of aryl nitriles (Scheme 5.81) [83], which are impor ant both in the pharmaceutical industry and in organic synthesis. In this cyanatio1 eaction, ammonium iodide and DMF were used as the source of nitrogen and carbo1 itom of the cyano unit, respectively (Scheme 5.82). It is assumed that DMF is ini ially oxidized to its iminium species A upon the reduction of Cu(II) salts to Cu(1) yanide ion is envisioned to form on the reaction of A with ammonia, presumabh 7ia an amidinyl species B. The reaction is believed to proceed via a two-step process nitial iodination and then cyanation. In the first step, the key intermediate iodoaren: ° is formed through the reaction of boronates with certain plausible iodide sources uch as NH,I or the in situ-generated Cul and I, where copper species facilitate it -onversion in the latter two cases. In the second step, cyanation of C takes plac yresumably upon the reaction with either cyanide anion under copper-mediatec](https://figures.academia-assets.com/35916932/figure_291.jpg)
![Copper-catalyzed C—H and C—X bond activations have attracted much attention because they can convert a C—H bond directly into a C—C or C—FG bond. These transformations meet the basic requirements of green chemistry [82]. One elegant example of a cascade reaction related with the copper-catalyzed C—H bond activation is the copper-catalyzed C—H oxidation/cross-coupling of indole with G-amino carbonyl compounds (Scheme 5.80).](https://figures.academia-assets.com/35916932/figure_292.jpg)





![In 2008, Sorimachi and Terada reported a relay catalysis using a rhodium hydride complex/Br¢gnsted acid (129) binary system (Scheme 5.88) [89]. The sequential transformation involves a three-step relayed catalysis, where (1) isomerization of allylamide 130 to enamide A is catalyzed by RhCIH(CO)(PPh,),; (2) subsequent isomerization of A to imine B is relayed by 129; and (3) the catalytic sequence is terminated by a carbon-carbon bond forming the reaction of B with a nucleophilic component 131 under 129. This approach enables the generation of reactive imines B from readily available allylamides 130 in a one-pot reaction via tandem isomerization. In 2009, Cai et al. reported a cascade olefin cross-metathesis/intramolecular Friedel-Crafts alkylation for the construction of polycyclic indoles 133 (Scheme 5.89) [90]. A Ru complex (134)/chiral Brgnsted acid [($)-135] binary system was used in this relay catalysis. Recently, the same group reported an enantioselective intramo- lecular aza-Michael addition of indoles using a similar binary catalyst system (Scheme 5.90) [91].](https://figures.academia-assets.com/35916932/figure_298.jpg)



![reduced yields. A mechanism involving two additions of organopalladium species to double bonds was proposed by the authors. Oxidative addition of the benzyl halide is followed by insertion of the less sterically hindered terminal alkene, affording alkylpal- ladium intermediate 3. Intramolecular insertion of the second alkene to the C—Pd bond is preferred over B-hydride elimination and generates five-membered ring 4 in a cascade fashion. Subsequently, the final product 5 would be afforded by B-hydride elimination and rearrangement of the newly formed double bond. The same group also reported a similar protocol to afford substituted cyclopentene compounds 7 by palladium-catalyzed cascade reaction of benzyl halides 2 with diethyl! diallylmalonate 6 [7b].](https://figures.academia-assets.com/35916932/figure_302.jpg)

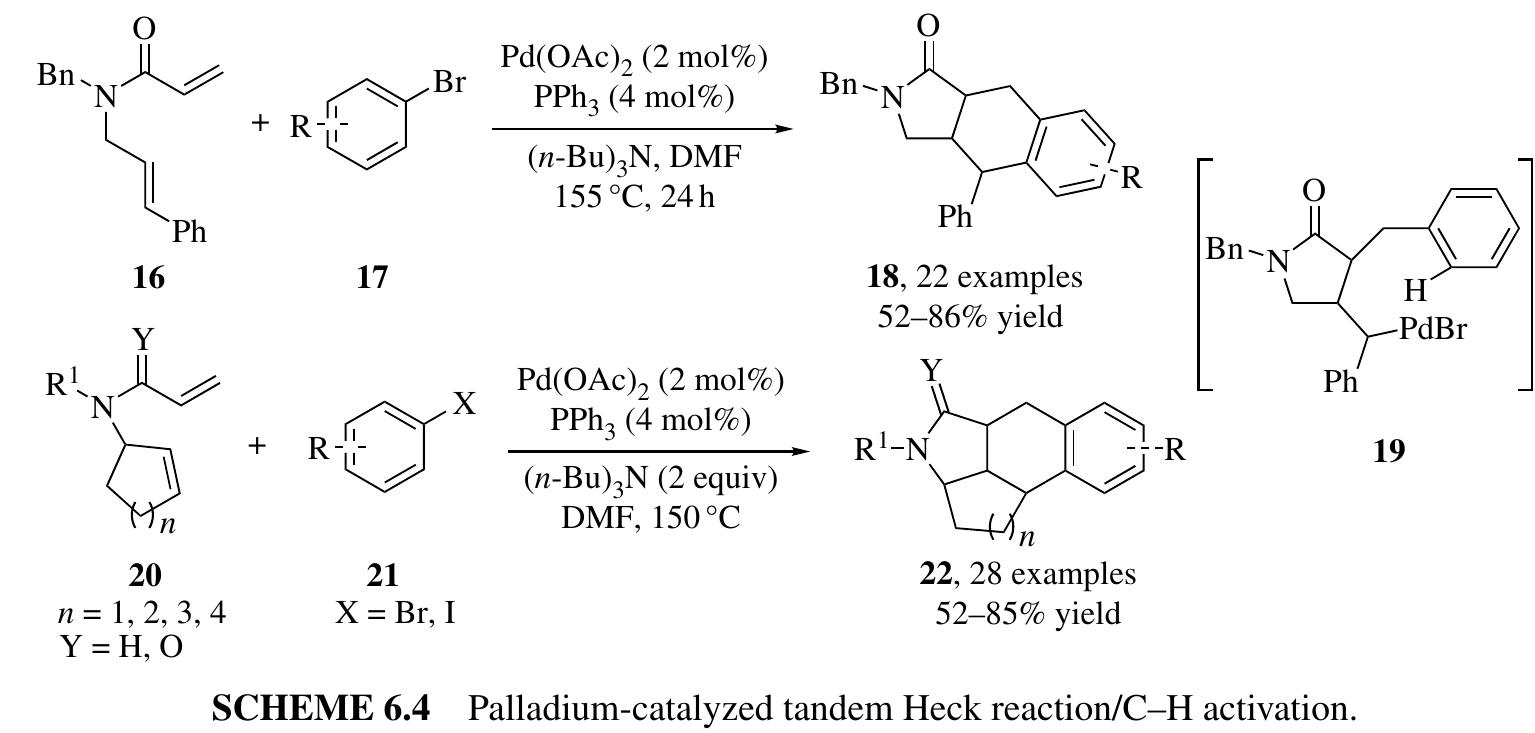

![SCHEME 6.6 C(sp*)—H activation/Heck cyclization/Heck arylation sequence. 6.2.3 Cascade Heck Reaction/Reduction/ Cyclization Felpin et al. developed a palladium-catalyzed tandem Heck reaction—reduction— cyclization synthesis of oxindoles (n=0) and dihydroquinolones (n= 1) using differ- ent 2-(2-nitrophenyl)acrylates (n=0) or 2-(2-nitrobenzyl)acrylates (n=1) and aryldiazonium salts 36 in good yield [15] (Scheme 6.7). However, a complementary approach was developed by the same group using diazonium salts 40 and substituted acrylates 41 to access a larger substitution pattern on the C3 position (Scheme 6.8). It is noteworthy that a different solvent was required and the charcoal had to be added after the Heck reaction. C3-unsubstituted, arylated, and alkylated dihydroquinolones 42 can be obtained efficiently using this protocol. The oxidative addition of iodobenzene to the Pd(O) catalyst in the presence of Ag,CO, would afford phenylpalladium species, followed by regioselective syn insertion to the C=C bond to generate the sulfonylalkylpalladium intermediate 26. Unlike the usual behavior of acyclic alkenes, the intermediate 26 would evolve faster through a C—H activation process, to afford the cyclopalladium species 27, than by B-hydrogen elimination to give the Heck product 24. The intermediate 27 could react further with iodobenzene through an oxidative addition/reductive elimi- nation pathway via a Pd(IV) palladacycle intermediate to form the o-alkypalladium intermediate 28. The repetition of the same sequence of steps to the second ortho C—H position would furnish the next o-sulfonylalkypalladium intermediate 30, followed by the third C—H activation process to afford the seven-member pallada- cycle species 31. Finally, the main product 25 would be afforded by the reductive elimination of intermediate 31. —s wmameaee Ee: . = 7 lw a: ‘ — * -](https://figures.academia-assets.com/35916932/figure_306.jpg)

![HEME 6.9 Palladium-catalyzed Heck cyclization/carbonylation cascade Accordingly, Pinto et al. developed an efficient synthesis of functionalized 3-alkyl-3-cyanomethyl-2-oxindole 50 by utilizing a palladium-catalyzed intramolec- ular domino Heck reaction/cyanation employing K,[Fe(CN),] as a trapping agent for the o-alkylpalladium intermediate [20] (Scheme 6.11). A wide range of substrates with different electronic properties could be introduced to the reaction. In addition, the authors accomplished a concise synthesis of physostigmine using this tandem process for the architecture of the core framework.](https://figures.academia-assets.com/35916932/figure_308.jpg)
![ound that simple triphenylphosphine is the best ligand for the reaction, and they ould obtain satisfactory results with as little as 0.1mol% catalysts. Furthermore, mly slight excess amounts of norbornadiene diester and boronic acid were required. Recently, Yahiaoui et al. developed a chelation-controlled palladium-catalyzed Teck—Mizoroki/Suzuki-—Miyaura domino reaction involving metal-coordinating limethylaminoethy] vinyl ethers and various electron-deficient and electron-rich rylboronic acids by the use of p-Bq as the oxidant [22] (Scheme 6.13). The two- arbon tethered dimethylamino moiety, which could combine with p-Bq for the tabilization of the o-alkylpalladium(II) intermediate 56 and formation of the dia- ylated products 57, is assumed to be crucial for avoiding the formation of Heck roducts 58. Lee et al. combined the intramolecular Heck reaction with Suzuki coupling and developed an efficient route to the preparation of 4-methylene-3-arylmethylpyrro- lidines [23] (Scheme 6.14). The authors didn’t observe direct Suzuki coupling bet- ween 59 and arylboronic acids. Interestingly, no B-hydride elimination occurs after double-bond insertion. Neighboring group participation of one of the oxygen atoms of the tosyl group depicted in 61 is assumed to stabilize the alkylpalladium intermediate and suppress the B-hydride elimination. Szlosek-Pinaud et al. developed a very simple and efficient method for the prepa-](https://figures.academia-assets.com/35916932/figure_309.jpg)




![SCHEME 6.18 Palladium-catalyzed tandem reaction for the synthesis of isoxazolidines. In 2010, Jaegli et al. reported a novel palladium-catalyzed intramolecular domino spirocyclization process for the preparation of biologically relevant spiropyrrolidine- 3,3’-oxindoles 86 [32] (Scheme 6.19). Oxidative addition of the aryl halide to Pd(0) aminopalladation via the coordinated intermediate 84 leads to palladacycle 85; reductive elimination of complex 85 generates the final product. Both Heck reaction and aminopalladation processes were viable pathways from amide 83, and the route that occurs is dependent on the ligand chosen. The use of BuMePhos as the ligand is required for the successful formation of spirooxindoles.](https://figures.academia-assets.com/35916932/figure_314.jpg)


![Early in 1992, Negishi et al. reported a palladium-catalyzed cascade involving alkynes toward benzene derivatives [38]. Recently, Blond et al. reported a palla- dium-catalyzed cascade reaction toward strained aromatic polycycles 100 [39] (Scheme 6.22). Bromoenediynes 98 undergo 4-exo-dig cyclocarbopalladation and subsequent 5-exo-dig cyclization to afford the vinylpalladium intermediate 99, which possibly undergoes a 6m-electrocyclization and a syn B-H elimination to furnish the products observed. Shibata et al. demonstrated an intermolecular three-component coupling of aryl or vinylhalides, diarylacetylenes, and monosubstituted alkenes, leading to the corresponding 1,3-butadiene or 1,3,5-hexatriene derivatives in the presence of palladium species [40] (Scheme 6.23).](https://figures.academia-assets.com/35916932/figure_317.jpg)


![Using the chiral cationic palladium(ID species as the catalyst, Yang et al. devel- oped highly enantioselective tandem [3+2] annulations of 2-acylaryboronic acids 113 with substituted alkynes to yield optically active 1-indenols 114 in high yields with excellent enantioselectivities [46] (Scheme 6.28). The vacant coordination site and high Lewis acidity of the palladium center in the intermediate 116 may activate the carbonyl group by coordination and make the nucleophilic addition occur easily. the carbonyl group by coordination and make the nucleophilic addition occur easily.](https://figures.academia-assets.com/35916932/figure_320.jpg)


![followed by Stille cross-coupling and 67 or 87 electrocyclizations [49]. Similar work was carried out by Kan and Anderson to prepare a series of fused bi- and tricycles [50] (Scheme 6.31). This transformation could tolerate carbon-, nitrogen-, or oxygen-tethered bromoenynes, which undergo a Heck-type reaction followed by Stille coupling with vinylstannanes to generate triene. Finally, the final products would be afforded in good yield by anin situ 67-electrocyclization. Just like Bour and Suffert’s work, this method can be applied for the syn- thesis of eight-membered cycles by employing dienylstannanes followed by 8n-electrocyclization. Fused tricycles can also be constructed efficiently using cyclic dienylstannanes. of a vinyltributylstannane reagent. The authors then summarized the study of differ- ent parameters of the reaction (catalyst, solvent, and temperature) and the starting materials, giving the optimal conditions to favor the cyclocarbopalladation process. These conditions were applied to realize 4- and 5-exo-dig cyclocarbopalladation followed by Stille cross-coupling and 67 or 87 electrocyclizations [49]. Similar work was carried out by Kan and Anderson to prepare a series of fused](https://figures.academia-assets.com/35916932/figure_323.jpg)



![Chen et al. explored a palladium-catalyzed synthesis of 3,4-disubstituted 2-trifluoro- methylquinolines 136 though a cascade Sonogashira coupling/alkyne carbocycliza- tion process from f-trifluoromethyl B-enamino ketones 135 with aryl-substituted alkynes [55] (Scheme 6.36). The authors suggested that this protocol includes Pd-catalyzed formal Sonogashira coupling, followed by base-mediated alkyne car- bocyclization of coupling product, and isomerization. This reaction affords the desired trisubstituted quinolines in good to excellent yields in just a few hours and under mild reaction conditions with high functional group tolerance. This reaction can be expanded to the non-fluorine-containing substrates. Polysubstituted benzenes are known useful compounds used widely in industry and academia. Thus, highly selective synthesis of polysubstituted benzene structures is in high demand. Although transition metal-catalyzed [2+2+2] cyclotrimerization of alkynes is the most frequently employed strategy for the preparation of polysubstituted benzenes, it suffers from serious chemo- and regioselectivity problems that lower the yield and dramatically affect the utility of the transformation. In 2005, Xi et al. developed a palladium-catalyzed one-pot multicomponent coupling reaction for the highly regiose- lective synthesis of polysubstituted benzenes [56] (Scheme 6.37). The Sonogashira coupling of terminal alkynes with vinyl bromides generates enynes 140, which readily undergo dimerization/benzannulation to afford polysubstituted benzenes 141. Both aryl- and alkyl-substituted alkynes could be introduced efficiently to this transformation. Huang et al. discovered an intriguing cascade reaction between 3-iodoenones](https://figures.academia-assets.com/35916932/figure_327.jpg)

![Recently, Wang et al. reported a novel palladium-catalyzed sequential Sonogashira/ carbopalladative cyclization/Suzuki reactions involving multiple carbon-carbon bond formation using protected homopropargyl alcohol 155 under mild conditions [59] (Scheme 6.40). Various indene derivatives 156 could be constructed efficiently with good yields in this transformation. Moreover, this reaction has a wide tolerance of various substituents in the substrates. Zhou et al. developed an interesting multicomponent reaction for the preparation of 1,2-dihydroisoquinolin-1-yl phosphonates 151 from readily available 2-bromo- benzaldehydes, alkynes, amines, and diethyl phosphite [58] (Scheme 6.39). The authors suggest that during the reaction process, 2-alkynylbenzaldehyde 152 would be afforded via palladium-catalyzed Sonogashira coupling reaction of 2-bromobenz- aldehyde with alkyne. The following condensation with amine would generate the o-alkynylarylaldimine 153, which would then undergo intramolecular electrophilic cyclization in the presence of a suitable Lewis acid. Subsequent nucleophilic addition of diethyl phosphite would afford the product desired.](https://figures.academia-assets.com/35916932/figure_329.jpg)

![In 2007, Pinto et al. developed a novel palladium-catalyzed three-component synthesis of 3-(diarylmethylene)oxindoles through a cascade Sonogashira/carbopalladation/C—H activation/ C—C bond-formation sequence from readily available N-aryl-N-alkyl propiolamides 160, aryl iodides, and a second aryl iodide [63] (Scheme 6.42). This is the first example of three different palladium-catalyzed reactions involving](https://figures.academia-assets.com/35916932/figure_331.jpg)
![SCHEME 6.43 Selective cascade carbopalladation/annulation reactions. EEA Me Tee Seen Le eS In 2010, Cherngak et al. developed an efficient methodology for the preparation of fused polycyclic indole derivatives 165 from readily available substrates 164 via a pal- ladium-catalyzed cascade carbopalladation/annulation reaction [65] (Scheme 6.43). The authors observed that the use of base has a dramatic effect on the selective inter- o1 intramolecular cyclizations leading to seven- or five-membered fused rings. If CsOAc was used as the base, direct intramolecular cyclization occured via C—H activation to afford five-membered fused rings 166. In contrast, seven-membered fused rings 165 could be formed when Et,N or KOAc was used as the base. Both electron-rich and electron-deficient alkynes could be introduced efficiently to palladium-catalyzed inter- molecular cyclization. Interestingly, with acyl-tethered substrates (X=CO), regardless of the base employed, the direct cyclization product 166 was afforded exclusively.](https://figures.academia-assets.com/35916932/figure_332.jpg)





![Curran and Du discovered an efficient route to fused tetracycles 180 via a palla lium-catalyzed cascade reaction of 6-iodo-N-propargyl-2-pyridones 179 and elec ron-rich aromatic isocyanides [73] (Scheme 6.50). The yield of this reaction i ‘easonable to good, but the substrate scope is limited to highly electron-rich aryl iso syanides. It is very important that this method can be used for the synthesis of the mos valuable classes of camptothecin and homocamptothecin analogs. DB-67, which ha sromising activity as an anticancer agent, could be synthesized efficiently by thi nethod. The authors proposed that the oxidative addition into the aryl iodide is th ‘irst step of this reaction. Vinylpalladium intermediate 181 was formed by isocyanid nsertion, followed by alkyne insertion in a C—Pd bond to generate intermediate 182 Finally, C—H activation of the aromatic ring results in the target product.](https://figures.academia-assets.com/35916932/figure_338.jpg)


![An interesting palladium-catalyzed cascade reaction leading to 3,3-disubstituted oxindoles 190 starting from 2-(alkynyl)aryl isocyanates 189 with benzylic alcohols has been developed by Toyoshima et al. [76] (Scheme 6.53). This reaction integrates a cyclization step and a novel [1,3] rearrangement step. Both benzylic and allylic alcohols could be introduced efficiently to this domino process. Furthermore, the products of this reaction are an important class of heterocycles which are often found in naturally occurring and biologically active molecules.](https://figures.academia-assets.com/35916932/figure_341.jpg)






![SCHEME 6.60 Palladium-catalyzed cascade aminopalladation/Heck coupling reactions. Gabriele et al. reported an unprecedented multicomponent domino reaction t functionalized indoles based on the combination between an initial nucleophili attack step on an imine moiety and a palladium-catalyzed oxidative heterocycliza tion/alkoxycarbonylation process [84] (Scheme 6.61). Initially, condensation o aldehyde and alkynylanilines 220 would generate 2-alkynylaniline imines 221 i the presence 4-A molecular sieves. Subsequent nucleophilic addition of ROH t the imine group of 221 followed by a Pdl,-catalyzed oxidative 5-endo-dig cycliza tion/alkoxycarbonylation would afford the final products 222 in moderate to goo yields.](https://figures.academia-assets.com/35916932/figure_348.jpg)
![Han and Lu used a cationic Pd(II)-catalyst system for the synthesis of 3-hydroxyme- thylindole 224 from readily accessible N-tosyl-2-phenylethynylanilines 223 and aldehydes [85] (Scheme 6.62). Intramolecular aminopalladation on the Pd(ID- coordinated alkyne generated the intermediate 225. Subsequent addition to the car- bonyl group of aldehyde afforded intermediate 227, followed by protonolysis to produce the products observed. This domino reaction provides an efficient way for the construction of functionalized indoles in one step in the presence of Pd(II) without the necessity of a redox system.](https://figures.academia-assets.com/35916932/figure_349.jpg)




![co So ee ee Le ae Recently, two groups have independently demonstrated that palladium-catalyzed cascade cyclization of enynes 246 under oxidative conditions would afford cyclopro- pyl fused y-butyrolactones [90] (Scheme 6.66). The reaction proceeds via a Pd(II)/ Pd(IV) catalytic cycle. Trans-acetoxypalladation of the alkyne moiety of 246 affords intermediate 249, followed by the intramolecular alkene insertion. The resulting Pd(II) species 250 will be oxidized by the PhI(OAc), to generate a Pd(IV) species 251, which would undergo cyclization via a S.2type of attack by the electron-rich alkene moiety tethered to the Pd(IV)-bound carbon with inversion of the configura- tion. The final product is obtained upon hydrolysis. Tsujihara et al. have also achieved the enantioselective variant of this transformation by use of a spiro-bis(isoxazoline) type of ligand and Pd(CF,CO,), [90c]. Using H,O, as a stoichiometric oxidant, Yin and Liu developed an efficient palla- dium-catalyzed oxidative cycylization of enyne 253 under mild conditions [91] (Scheme 6.67). This transformation is believed to proceed through a mechanism involving oxidation of sp*> C—Pd(II) species by H,O,, and the resulting sp’ C—Pd(IV)-Cl intermediates undergo direct reductive elimination to give the final product with the retention of the configuration at the carbon center. In 2009, Ye et al. developed an efficient protocol for the preparation of 3-chloro- and 32-hraqamo-_1-methvleneindenes 9564 via a nalladinm-catalvzed damingo reaction](https://figures.academia-assets.com/35916932/figure_354.jpg)




![Yoshida et al. uncovered a novel palladium-catalyzed cascade reaction of propar- gylic carbonates with phenols which involves a CO, elimination/fixation step and affords phenoxy-substituted cyclic carbonates. They discovered that this reaction proceeds in a highly enantiospecific manner to give the chiral cyclic carbonates 276 and 277 via an overall cascade chirality transfer process through use of the chiral propargylic carbonates 275 as the substrates [98] (Scheme 6.73). In 2008, Ohno et al. introduced two nucleophiles to the propargyl bromides, struc- tured as 1,7-diamino-5-bromohept-3-yne derivatives 278. These compounds were treated with catalytic Pd(PPh,), in the presence of NaH in MeOH. The reactions give 2,7-diazabicyclo[4.3.0]non-5-enes 280 in good yields, in which the regioselectivity of the reaction is controlled completely by the relative reactivity of the amine functional groups, irrespective of the position of the nucleophiles [99] (Scheme 6.74).](https://figures.academia-assets.com/35916932/figure_359.jpg)






![Grigg etal. combined alkene insertion with nucleophilic attack and 7-allylpalladium species and developed a successful procedure for the preparation of spirocycles 313 [108] (Scheme 6.81). Aryl iodides with a tethered nucleophile structured as 311 and allenes were employed as starting materials. The nucleophiles can be amines or malonates, and simple allene or dimethylallene can be used. Various substituted spirocycles 313 could be obtained in reasonable yields.](https://figures.academia-assets.com/35916932/figure_366.jpg)
![SCHEME 6.79 Palladium-catalyzed carbopalladation/allylic alkylation domino sequence. activation onto the aromatic ring to afford tricyclic product 302 in a single step. In 2009, Kemmerer et al. uncovered a phosphine-free carbopalladation/allylic alkylation cascade sequence for the synthesis of 4-(a-styryl) y-lactams 308 [106] (Scheme 6.79). The reaction pathway of this transformation involves the formation of m-allylpalladium(ID species 307, which was trapped by the intermolecular active methylene. Both electron-rich and electron-deficient aryl iodides could be intro- duced efficiently to this cascade process. Li and Dixon developed a stereoselective and efficient protocol for the synthesis of spirolactam 310 employing a similar carbopalladation/z-allvlnalladium trapping strategv [107] (Scheme 6.80).](https://figures.academia-assets.com/35916932/figure_367.jpg)
![SCHEME 6.83 Palladium-catalyzed cascade carbon monoxide insertion reaction. Grigg et al. reported a successful four-component domino reaction for the syn- thesis of functionalized dienes 316 from ary] iodides, allyl amine derivative, allene, and carbon monoxide [110] (Scheme 6.83). Carbon monoxide could insert into the C—Pd bond of arylpalladium(ID) iodides to generate a carbonylpalladium species, which is followed by allenylation to form 7-allylpalladium species. Finally, the attack of the nitrogen nucleophile produces the product observed. The products of this domino multicomponent reaction could be subjected efficiently to ring-closing metathesis in the presence of Grubbs’ second-generation catalyst.](https://figures.academia-assets.com/35916932/figure_368.jpg)
![In 2002, Ma et al. developed an efficient route to polysubstituted cis-pyrrolidin Jerivatives 325 through a palladium-catalyzed three-component tandem double-addition syclization reaction [113a] (Scheme 6.86). Initially, carbopalladation of allene woul senerate 1-allylpalladium species 326, followed by nucleophilic addition to imine: iffording intermediate 327. Subsequent nucleophilic attack of nitrogen to 1-allylpalladiur would produce the 5-exo cyclization product 325 and regenerate the active catalyst. Th mechanism was verified by mass spectral methods [113b]. Later, the same grou Jeveloped a similar three-component reaction by using the dibenzyl azodicarboxylat nstead of the tosyl imines, providing efficient access to enantioenriched pyrazolidin Jerivatives under the catalysis of a palladium and copper catalyst [113c]. Palladium-catalyzed three-component reactions of electrophiles, allenes, an](https://figures.academia-assets.com/35916932/figure_369.jpg)
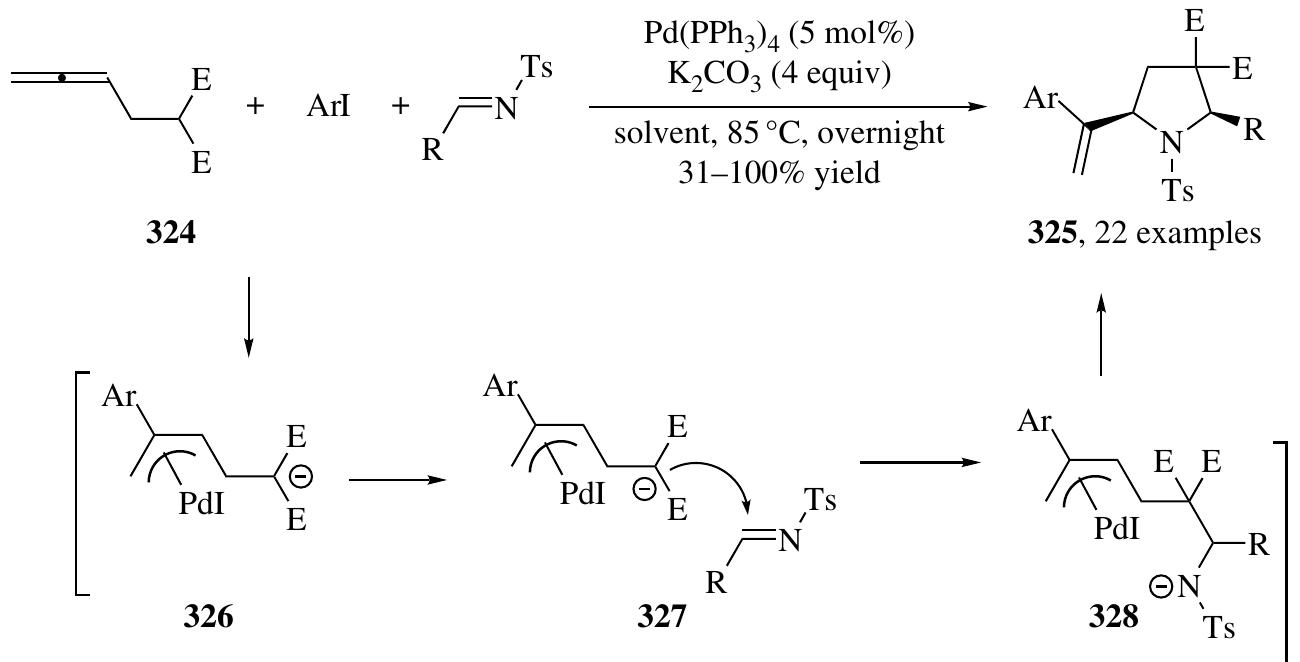

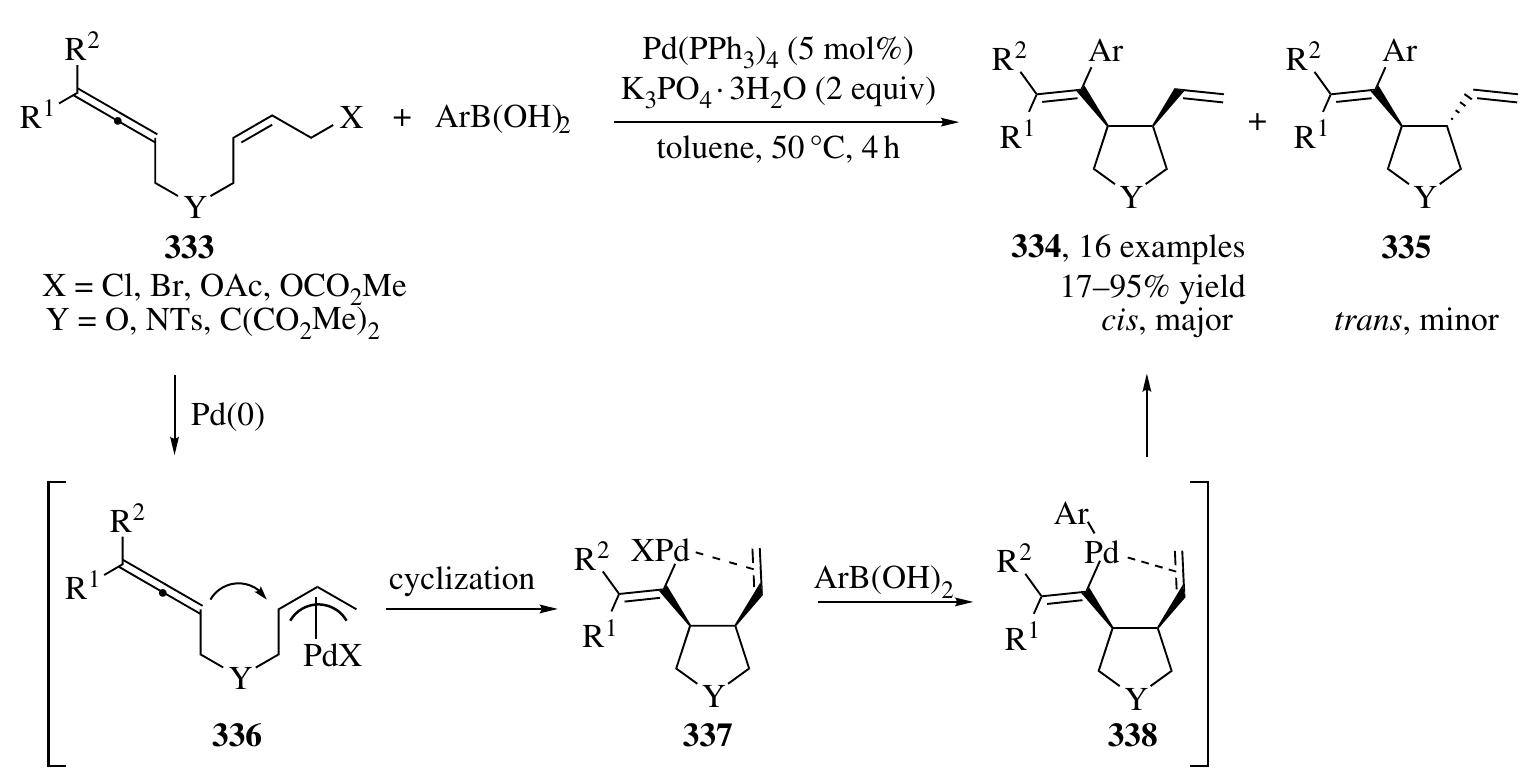




![SCHEME 6.94 Palladium-catalyzed cyclization of 2,3-allenoic acids. Since 2002, Ma et al. have demonstrated the cross-coupling/cyclization of 2,3-allenoic acids 346 in the presence of other functionalized allenes, such as 1,2-allenyl ketones 368 {121] or 2,3-allenols 370 [122], affording differently substituted 2(5H)-furanones 369 and 371. They also studied the cascade cyclization/cross-coupling reaction of 2,3-allenoic acids in the presence of simple monosubstituted allenes, which yielded the stereodefined 4-(bromo-2(£)-alken-2-yl)-2(5H)furanones Z-372 [123] (Scheme 6.94).](https://figures.academia-assets.com/35916932/figure_377.jpg)




![cis-Carbopalladation, anti-oxopalladation, and reductive elimination to afford the final tricyclic products 382 has been proposed by the authors [125] (Scheme 6.96). In 2009, Shu et al. reported a successful palladium-catalyzed three-component cas- cade cyclization reaction of propargylic carbonates 386 with 1,5-bisallenes 387 and organoboronic acids for the efficient preparation of cis-fused bicycle[4.3.0]nonenes 388 [126] (Scheme 6.97). The bisallenes can be nitrogen- or carbon-tethered [X=NTs or C(EWG),] and the alkynyl carbonates can have aryl or alkyl substituents (R'=Ph or n-Bu). It is noteworthy that the final products of this cascade reaction exist in some bioactive molecules, such as the analogs of asparvenone. The authors proposed that this reaction may involve three carbopalladation reactions to afford sequentially a t-allylic palladium 390, a vinylic palladium 391, and a novel 7-allylic palladium intermediate 392. The final step is a Suzuki coupling reaction of the 1-allylic palla- dium intermediate 393 with organoboronic acids to afford the observed product.](https://figures.academia-assets.com/35916932/figure_382.jpg)
![considered by native populations to possess medicinal properties. It is used to improve digestion and to protect the stomach in Paraguay, to cure hypertension in Taiwan, and to treat toothaches, blennorhagia and stomach disorders in India [4]. Overman et al. reported a very unique strategy to construct this molecule (Scheme 7.1) [5]. The key construction unit is the [3,2,1] bridge core, including the sterically congested, bridged bicyclic system and two quaternary stereocenters. Overman et al. envisaged that both the two rings and the two quaternary centers could be created in a single cascade Heck reaction. As illustrated in Scheme 7.1, treatment of substituted cycloheptene 1 with Pd(OAc), (10mol%), PPh, (20 mol%), and Ag,CO, in THF generated intermediate 2 through the initial oxidative addition into the carbon—iodine bond. Then, the first intramolecular Heck reaction occurred to generate alkylpalladium(ID intermediate 2, while B-hydride elimination did not hap- pen in the absence of suitable hydrogen atoms. Subsequently, the second 1,2-inser- tion reaction occurred on the trisubstituted alkene. The intermediate 3 was formed, followed by B-hydride elimination to afford product 4 in 90% yield as a single stereoisomer. This successful strategy demonstrated the synthetic power of the intra- molecular Heck reaction for the generation of tertiary and quaternary stereocenters (in both a diastereo- and enantioselective fashion) and multiple-ring systems even in sterically crowded environments. Estrone is an estrogenic hormone. Tietze’s group reported a very elegant method for the enantioseletive total synthesis of estrone (Scheme 7.2) [6]. The best part of the strategy is the generation of the steroid ring B through consecutive inter- and intramolecular Heck reactions. They discovered that the treatment of functionalized aromatic compound 5 and the enantiopure hydrindene derivative 6 with Pd(OAc), and PPh, in the presence of n-Bu,NOAc in a mixed DMF/MeCN/H,O](https://figures.academia-assets.com/35916932/figure_383.jpg)
![YUAaHULAUVe Yield, In 2002, Tietze’s group used the similar strategy to synthesize structurally simpli- fied cephalostatin analogs by a multiple Heck reactions (Scheme 7.3) [7]. The first step is a selective Heck reaction of hydrindene 11 with 6 at the vinyl bromide moiety, followed by a sequence of a Corey—Fuchs reaction and selective debromination. Next, compound 14 reacted with 6 to yield the desired diindenylethenylbenzene 15 through an intermolecular Heck reaction in 47% yield in a stereo- and regioselective way. The following intramolecular Heck reaction of 15 occurred with a catalytic amount of palladacycle 12 at 130 to 140 °C for 1.5h to generate 16 in 80% yield. The conversion proceeded with high selectivity and led to the exclusive formation of the multiple cyclic compound.](https://figures.academia-assets.com/35916932/figure_384.jpg)


![Ge 4il BEM Sony ted SAR Set Met Be SD J Bae ATE ® Maddaford et al. reported an interesting palladium-mediated “domino” polyene yclization containing asymmetric Heck reaction to synthesize halenaquinone- elated natural product (+)-xestoquinone (Scheme 7.5) [14] which is the first appli- ation of the asymmetric palladium-catalyzed polyene cyclization directed toward he synthesis of a natural product. This well-orchestrated sequence of events began vith the treatment of ary] triflate 20 with Pd(OAc),, S-BINAP, and PMP in toluene, ind the oxidative addition of a Pd(0) species into aryl triflate 20 initially generated | Pd(ID) complex. At this time the chiral BINAP ligand remained anchored to the netal center through both the alkene coordination event and subsequent 1,2-inser- ion step to generate o-alkylpalladium(II) intermediate 21, which ensured a higher evel of enantioselectivity. Then, intermediate 21 underwent another migratory nsertion reaction followed by B-hydride elimination to generate pentacyclic ompound 22 with a respectable 68% ee and regenerated the palladium catalyst. iven though the enantioselectivity of this reaction was not optimal, it was the first pplication of an asymmetric palladium-catalyzed polyene cyclization directed](https://figures.academia-assets.com/35916932/figure_387.jpg)
![Heck reactions on nonconjugated dienes create an electrophilic m-allyl complex that is susceptible to nucleophilic attack. When the compound contains a conjugated diene, the regioselectivity becomes a very important issue, which must be considered. Cascade sequence involving a Heck reaction and a r-allyl reaction could be found in the formal total synthesis of Morphine by Overman [15]. Treatment of dienes with Pd(OCOCF,),(PPh,), and NEt, in toluene led to formation of the backbone of Morphine in 56% yield (Scheme 7.6). In 2000, Overman and Rosen reported another example of the total synthesis of (—)-Spirotryprostain B [16] using the same principle (Scheme 7.7). In contrast to the example described above, the quaternary spiro and adjacent stereocenters were constructed by adding an appropriate chiral ligand. Controlled by R-BINAP, 7?-allylpalladium intermediate 28 was generated through a suprafacial intramolec- ular Heck reaction of compound 27 in a favored 5-exo sense. Subsequently, intermediate 28 was trapped by nitrogen in tethered diketopiperazine to furnish the desired spiro product 30 in 26% yield. Compound 27 contains a conjugated triene; therefore, the intramolecular Heck insertion of conjugated triene could proceed with high regioselectivity.](https://figures.academia-assets.com/35916932/figure_388.jpg)



![Over the past decades, one of the most important families of natural products has attracted significant attention from synthetic chemists. Their structures contain a cyclic es Rapamycin, isolated from Streptomyces hygroscopicus found in an Easter Islanc soil sample [21], has potent antibiotic, cytotoxic and immunosuppressive activity [22]. Rapamycin contains a 31-membered ring, a plethora of asymmetric and geomet- rical centers, and sensitive functionality which poses a formidable challenge tc synthetic chemists. In 1993, Nicolaou et al. achieved the total synthesis of rapamycir (Scheme 7.10) [23]. In this process, the most impressive reaction was the double Stille reaction of vinyldistannane 42 with the corresponding bis(viny1 iodide) 40, whict resulted in the simultaneous construction of the sensitive conjugated triene system anc the macrocyclic skeleton 41. It is worth noting that the precursor 40 involved in thi: cascade has no protecting groups. This new strategy provided a highly valuable route for the construction of macrocycles in natural product total synthesis.](https://figures.academia-assets.com/35916932/figure_392.jpg)
![enediyne, and all of these compounds are potent antitumor agents. Shair et al. reported the total synthesis of the enediyne anticancer antibiotic dynemicin (Scheme 7.11), in which a double Stille reaction was employed to construct the structure of cyclic enediynes [24]. The substrate did not participate in a tandem Sonagashira reaction, which suggested that a double Stille coupling reaction was the only remaining route to accomplish this cyclization. The conversion of 42 to 43 was accomplished by using N-iodosuccinamide (NIS). The bis(iodoalkyne) substrate 43 reacted smoothly with cis- 1,2-distannyl ethylene 45 in the presence of a catalytic amount of [Pd(PPh,),] in DMF at 75°C to furnish the strained 10-membered-ring product 44, in a remarkable 81% yield. However, special attention must be paid in this cascade process. First, to avoid intermolecular dimerzation, the reaction had to be carried out at a relatively low dilu- tion (0.05 M). Second, if the epoxy group was substituted for by other groups, a Stille coupling reaction failed to proceed. In view of this, we should point out that the subtle conformational effect played a deciding role in this process. , 2, a |e, ee A en ee a Pee tne | Vee ee ey reer: 2. a: en a |e](https://figures.academia-assets.com/35916932/figure_393.jpg)
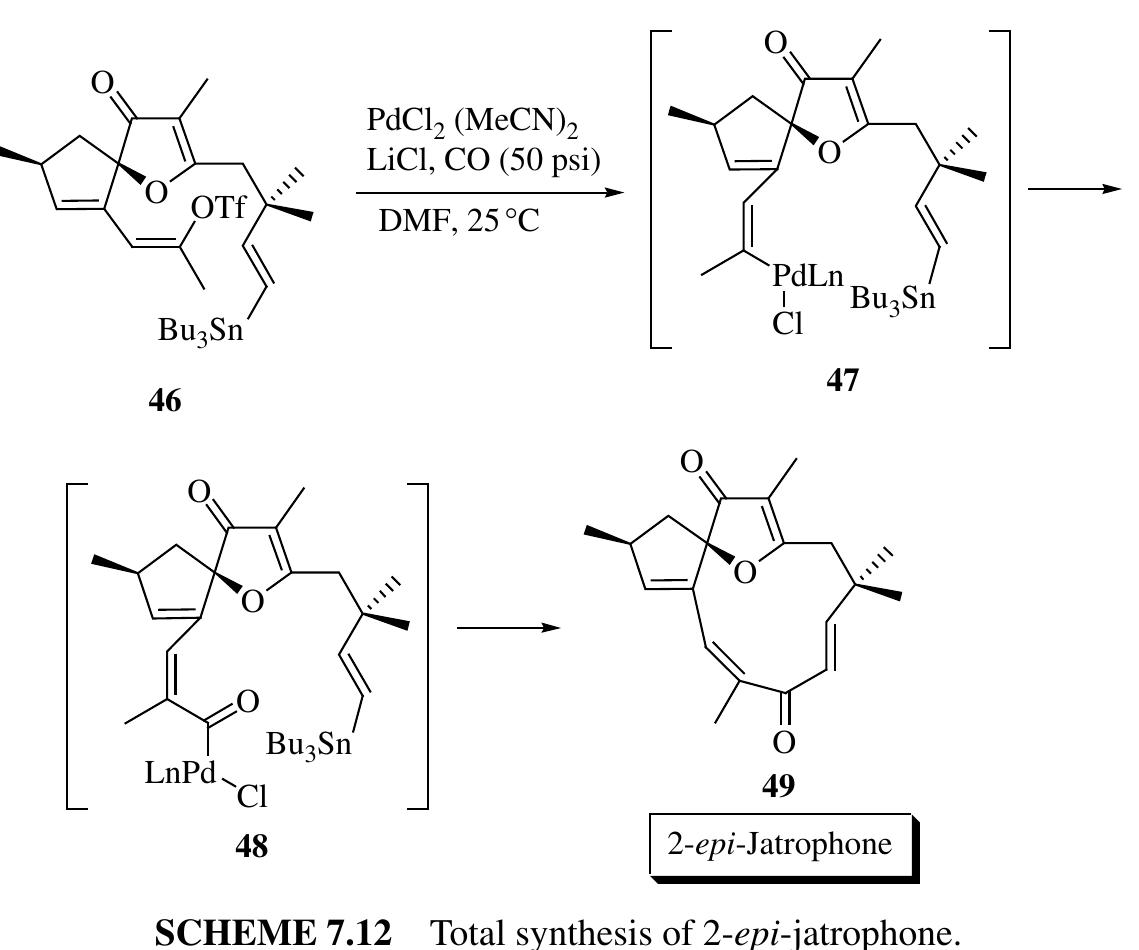



![Panepophenanthrin was isolated from the fermented broth of the mushroon strain Panusrudus IFO8994 in 2002 by Sekizawa et al. [29]. It is the first knowr inhibitor of the ubiquitin-activating enzyme (E1), which is indispensable for the ubiquitin—proteasome pathway [30]. As outlined in Scheme 7.15, the overall bio. mimetic strategy was employed in the total synthesis. A retro Diels—Alder revealec hat panepophenanthrin 60 could be disconnected to give 2 equiv of the knowr conjugated diene 61, and 61 could be formed from vinyl stannane 65 and viny bromide (bromoxone) 62 by a Stille coupling reaction. It was demonstrated tha his transformation could be accomplished by treatment of vinyl stannane 65 anc vinyl bromide (bromoxone) 62 with Pd,(dba), and AsPh, in toluene at 105 °C [31]. This cascade involved a Stille coupling reaction, a hemiketal forming and <](https://figures.academia-assets.com/35916932/figure_398.jpg)




![isa eee see BEY Be The Tsuji—Trost reaction, which involved the insertion of 1-allylpalladium species into alkenes, is a very useful method for the formation of five- and six-membered car- bocyclic and heterocyclic ring systems. Oppolzer et al. developed a cascade sequence combining palladium-catalyzed allylation and carbonylation reaction to synthesize the heteroyohimbine alkaloid 3-isorauniticine 97 (Scheme 7.21) [44]. Treatment ot compound 92 in AcOH with a catalytic amount of [Pd,(dba),] (10 mol%) and P(n-Bu). (30mol%) at 80°C under carbon monoxide (1 atm) initiated an allylation and carbon- ylation reaction cascade, which gave the bicyclic enone 96 in a reproducible yield ot roughly 50%. The first step in this sequence is the formation of 1-allylpalladium intermediate 93 by catalyst Pd(O). Then the Tsuji—Trost reaction occurred to generate the corresponding 1'-alkylpalladium(II) intermediate 94. The insertion of carbor monoxide into a C—Pd bond generated the next intermediate 95. Finally, a Heck-type reaction happened, to afford the product 96 in 48% overall yield.](https://figures.academia-assets.com/35916932/figure_403.jpg)
![(Scheme 7.22). In this total synthesis, a Pd-catalyzed asymmetric allylic alkylation was employed as the key step. However, the best enantioselectivity achieved in this key step using (S)-BINAPO 105 was only 54% ee, with 103 produced in 30% yield. In 2006, Chapsal and Ojima used novel chiral biphenol-based monodentate phos- phoramidite ligands and accomplished total synthesis with 99% ee in 41% overall yield [46]. Starting from the alkylated product 100, another two rings of this natural product were formed through an intramolecular allylic alkylation/Heck reaction cas- cade sequence. Treatment of 100 with Pd(OAc), (5 mol%), dppb (10 mol%) and NaH in DMF at 50°C triggered the initial intramolecular allylic alkylation to generate the intermediate 101, which underwent the anticipated intramolecular Heck cyclization by addition of i-Pr,NEt at 100°C to afford the key skeleton 103 in 58% yield as a single diastereoisomer.](https://figures.academia-assets.com/35916932/figure_404.jpg)
![a I I A Peixoto et al. reported a novel cascade reaction to complete formal asymmetric synthesis of Echinopine A and B (Scheme 7.23) [49]. Through the retrosynthetic analysis, [5,6,7] tricycle 109 is a key intermediate for this synthesis. They envisaged that transition metal—mediated cycloisomerization/intramolecular Diels-Alder may be followed spontaneously by an intramolecular Diels—Alder reaction to construct the [5,6,7] tricycle ring system. The proposed cycloisomerization happened smoothly by the treatment of 107 with Pd(OAc),/PPh, at an elevated temperature (80°C) to give diene enoate 108. Then, 108 participated in a subsequent intramolecular Diels— Alder reaction upon prolonged heating at higher temperature (160°C) to furnish [5,6,7] tricycle 109 in 75% overall yield from 107.](https://figures.academia-assets.com/35916932/figure_405.jpg)


![In 2004, Quinn et al. reported the total synthesis of (—)-Muricatacin (Scheme 7.27) [61]. The key synthetic step in this approach is a tandem ring-closing/cross-metathe- sis reaction. Acyclic trieneslactone 118 was used as the starting material, and the ring formation and chain extension were achieved by a tandom olefin metathesis. Ring- closing metathesis of 118 would provide an intermediate butenolide 119, in which the two olefins are clearly differentiated by their electronic environments. An inter- molecular cross-metathesis between the terminal olefin of 119 and an alkene cou- pling partner completed the tandem, to provide an extended hydroxyalkenylbutenolide 120. Quinn et al. found that the exposure of acyclic trieneslactone 118 with catalyst 122 (10mol%) in CH,Cl, gave 120 in 57% yield. The reason for the low yield is the decomposition of catalyst 122. To maintain a constant concentration of active cata- lyst, 122 was added in a 0.01 M solution over 8h via a syringe pump. With this mod- ification, the yield increased to 65%. Followed by reduction of the olefins and removal of the benzyl protecting group by catalytic hydrogenation or hydrogenoly- sis, the total synthesis of (—)-Muricatacin was completed. Another important cascade process involves ring-opening metathesis. Ring-opening metathesis polymerization (ROMP) is a type of olefin metathesis polymerization that produces industrially important products. In the context of total synthesis, many cascade sequences triggered by the initial ring-opening metathesis of a strained ring system have been developed. Among these, ring-opening/ring-closing metathesis (ROM—-RCM) processes involving bicyclo[2.2.1]heptenes were studied extensively for the synthesis of all-carbon and heteroatom-containing [.3.0]bicyclic systems. OWne anch annlicatiann ic the recent tatal eunthece af Ahnratnhnalactam A hy Henderecan](https://figures.academia-assets.com/35916932/figure_408.jpg)
![Hart and Philips made use of this type of tandem methylenation/ring-closing metathesis cascade sequence in their recent total synthesis of (+)-Cylindramide A 130 (Scheme 7.29), which features the use of a novel ring—opening/ring—closing—cross— metathesis (ROM—RCM-CM) strategy to build the characteristic bicyclo[3.3.0] octene ring system of the natural product [63]. They found that treatment of the nor- bornene 127 with 4mol% Grubbs’ catalyst in the presence of 3.0 equiv of 128 gave 129 in 59% yield through tandem ROM-RCM-CM reaction as a 2:1 mixture of separable diastereoisomers. With this cascade reaction completed, the total synthesis of (+)-Cylindramide A could be achieved in just a few steps.](https://figures.academia-assets.com/35916932/figure_409.jpg)

![More recently, Phillips showed that the analogous bicyclo[2.2.2]octene car participate in such a metathesis sequence to produce an [n.4.0] bicyclic system including the cis-fused decaline structure [64]. A beautiful early example of this type of protocol can be found in the expeditious synthesis of (+)-Lepadin B by Barbe anc Charette [65]. As shown in Scheme 7.30, treatment of readily available 131 with the second-generation ruthenium catalyst 122 (2mol%) in toluene at 80°C effected its smooth conversion into the corresponding bicyclical compound 132 with complete transfer of chirality from the original bicycle[2,2,2]octene to the newly formed bicy- clical system. It is noteworthy that rigorous exclusion of air and moisture was not required for this process, and adding the catalyst portion-wise was beneficial anc particularly crucial for reliability on a multigram scale. Pfeiffer and Phillips employed this process in their recent total synthesis of (+)-Cyanthiwigin U (Scheme 7.31) [66]. Exposure of 134 to catalyst 122 under an atmosphere of ethylene provided tricycle 135 in 43% yield through three steps, which established a concise route to the carbocyclic skeleton of the cyanthiwigin 136 in one pot. Two general mechanistic pathways could explain the formation of 135 from 134: (1) ring-opening metathesis of the bicyclo[2.2.2]octene leads to intermediate 137 or 138, which can subsequently undergo ring-closing metathesis to provide 135; and (2) initial metathesis of the endo enone, followed by reaction with the olefin of the bicyclo-[2.2.2]octene, leads to metallacyclobutane 139, which undergoes ring opening to yield 140. Subsequently, 140 is converted to 135 through a ring-closing metathesis. Raprdblescwwwmatvecnsd Aj] Taxed hae DD bamlkiocedlinc weenie ead =e same: a ws ook eee: ere ST](https://figures.academia-assets.com/35916932/figure_411.jpg)




![SCHEME 7.35 Total synthesis of (+)-B-Erythroidine. Oe Recently, the Erythrina alkaloids have aroused more and more concern, due to their intriguing biological activity and characteristic polycondensed structures, which pose significant challenges for synthetic chemists. Hatakeyama et al. devel- oped an efficient method for the construction of the erythrinan skeleton, which is based on a cascade ring-closing metathesis reaction, later employed in the total syn- thesis of (+)-B-Erythroidine 168 (Scheme 7.35) [76]. Specifically, treatment of 163 with thionyl chloride in pyridine resulted in dehydration to give an inseparable mixture of o,B-unsaturated lactone 164 and B,y-unsaturated lactone 165 (74:26). Then treatment of this isomeric mixture with 0.1 equiv of Grubbs I catalyst 125 in CH,Cl, at room temperature for 6.5h gave (+)-B-erythroidinein 168 in 42% yield together with 167 (4%) accompanied by an appreciable amount of unconverted 165 (31%). In this particular case, Grubbs II catalyst 122 turned out to be less effective and gave 168 in less than 30% yield.](https://figures.academia-assets.com/35916932/figure_416.jpg)
![Cochleamycin A, isolated in 1992 from a cultured broth of Streptomyces DT 136.1, has significant antimicrobial activity against gram-positive bacteria [77] ind cytotoxicity against P388 leukemia cells (IC,,, 1.6g/mL) [78]. The 5,6-fused ind 10,6-bridged tetracyclic core structure attracted much attention, which led to a 1umber of impressive synthetic studies [79]. Mukherjee and Lee developed a andem dienyne ring-closing metathesis of alkynyl silaketal to establish the (£,Z)- |,3-diene moiety required for a Diels—Alder reaction in the presence of Grubbs II -atalyst and achieved formal total synthesis of (—)-Cochleamycin 173 (Scheme 7.36) 80]. Basically, bicyclointermediate 170 was formed when silaketal 169 was >xposed to Grubbs II catalyst in DCE at 80°C. The removal of the silicon ether hrough protodesilylation allowed for the generation of stereochemically defined | .4-substituted (E,Z)-1,3-dienes 172. A very powerful cascade reaction had been developed by Cho and Lee i their approach to the total synthesis of (3R,9R,10R)-Panaxytriol 17 (Scheme 7.37) [81], which was isolated from Panax ginseng in 1983 [82]. Th cascade sequence was initiated by relay metathesis, which is then followed b metallotropic [1,3]-shift and cross-metathesis. This approach has become a efficient way for the synthesis of natural products with highly unsaturate carbon skeletons. Treatment of 174 with Grubbs second-generation catalyst i CH,Cl, at 40 °C in the presence of 2.0 equiv of alkene 175 generated the expecte product 178 in 61% yield as a mixture of Z/E-isomers. Surprisingly, rutheniur alkylidene 176 was isolated in 10% yield and could be converted to 178 upo treatment with 175. This confirms that complex 176 is a catalytically viabl intermediate in the catalytic cycle. Metathesis has arguably influenced the landscape of synthetic organic chem](https://figures.academia-assets.com/35916932/figure_417.jpg)


![ail With appropriate functional groups on the alkene and alkyne partners, this cou pling reaction can form substituted butenolides in one step. Trost and Miille employed this process in their total synthesis of the (+)-Ancepsenolide (Scheme 7.40 [85]. Treatment of alkene 192 and alkynoate 191 with 5mol% Cp(cod)—RuCl i methanol at reflux produced two products, 198 and 199, in a 2.9:] ratio in 51% isc lated yield with 198 as the major product. The mechanism was shown in Scheme 7.4( First, ligand exchange and coordination of the alkene and alkyne component occurred to afford 194, followed by oxidative cyclization to generate ruthenacyclo pentene 195. Then intermediate 196 was obtained by syn-B-hydride eliminatior which underwent reductive elimination to yield the product observed, 197, alon with regeneration of the catalyst 193. Diene 197 underwent lactonization to giv butenolide 199 as the desired product.](https://figures.academia-assets.com/35916932/figure_420.jpg)
![HUI SUO WIE AluULe aU d LOU Sel Ledeuoll UIE, Recently, Ma’s [87] and Echavarren’s [88] groups have reported the total syn- thesis of (+)-Englerin A and B by using gold catalysis cascade reaction independently (Scheme 7.41). Ma’s group used AuCl as the catalyst and produced the oxatricyclic derivative in 48% yield. Echavarren’s group adopted [IPrAuNCPh]SbF, as the cata- lyst and produced the oxatricyclic derivative 207 in a 58% yield. This cascade sequence is believed to proceed as shown in Scheme 7.41. First. enynes 200 reacted](https://figures.academia-assets.com/35916932/figure_421.jpg)
![HIPIe DULCE DIOCKS, The transition metal-catalyzed formation of isobenzopyrylium salt with a subsequent Diels—Alder reaction was proven to be a very valuable method for the onstruction of highly functionalized carbocyclic ring systems. Heliophenanthrone las a tricyclic ring system and is a suitable target for this cascade process. Dyker and dildebrandt adopted this strategy to the total synthesis of Heliophenanthrone Scheme 7.42) [89]. Exposure of dialkynyl ketones 210 to gold(IID chloride led to he sequence desired, giving tricyclic compound 213 in a 35% overall yield. By hanging to platinum(II) chloride as the catalyst in dioxane at 120°C, the overall yield of 213a/b was raised to 71%, the mechanism of which involves an isobenzo- yyryliumcation intermediate 211, which is formed by nucleophilic attack of the -arbonyl oxygen on the alkyne. The intermediate 211 then reacted with another ilkyne to give the TBDMS-protected heliophenanthrone 213. Following excision of he TMDMS group using aqueous HF in acetonitrile completed this total synthesis. The transition metal-catalyzed enyne cycloisomerization is one of the most impor- tant strategies for the synthesis of functionalized cyclic structures [90]. The reaction has been applied widely in the total synthesis, since polycyclic structures could be nicely assembled from acyclic precursors in a single step. A number of transition metal complexes are capable of catalyzing enyne cycloisomerizations. Among them,](https://figures.academia-assets.com/35916932/figure_422.jpg)
![gold and platinum complexes are especially powerful, which can deliver a diverse array of cyclic products under mild conditions with excellent chemoselectivity and high atom economy [91]. Simmons et al. found a rapid way to build a polycyclic system by using a GaCl,-catalyzed enyne cycloisomerization reaction as a key step. Later they applied this method to the total synthesis of several icetexane diterpenoid natural products [92]. However, when they applied the method to the synthesis of cortistatin pentacyclic core structure, they found that cleavage of the TBS ether hap- pened as a competing side reaction during the treatment of indene 215 with GaCl, (Scheme 7.43) [93]. The authors also reported that use of the platinum-based catalytic system avoided this problem, which proceeded in essentially quantitative yield to give the target molecule. The process started by chemoselective metal complexation to the alkyne, followed by cyclopropanation of the proximate alkene to produce cyclopropyl metal carbine 217. Then two consecutive 1,2-alky] shifts produced zwitterionic com- plex 218. Opening of the cyclopropyl ring and elimination released the catalytically active species and product 219. The utility of the platinum-catalyzed enyne cycloisomerization for the formal evnthecis of Rasenhilin whichis 9 memher of nrodisinine familv of alkaloids. was](https://figures.academia-assets.com/35916932/figure_423.jpg)



![SCHEME 7.48 Synthesis of the hexahydropyrrolo[2,3-b]indoline skeleton using the CRI reaction.](https://figures.academia-assets.com/35916932/figure_427.jpg)


![Ce gh OES An intermolecular cycloaddition cascade was also adopted in total synthesis. Nakamura et al. reported the total synthesis of zaragozic acid C by using this sequence (Scheme 7.51) [108]. When a solution of the 6-tartrate-derived o-diazoester 247 was added slowly to a mixture of alkyne 248 (3.0 equiv) and Rh(II]) acetate dimer (5mol%) in refluxing benzene, the bicyclic compound 250 was formed as a single stereoisomer in 72% yield. The stereoselectivity of this reaction is dictated by approaching the alkyne dipolarophile to the top face of carbonyl ylide intermediate 249 to avoid steric interactions with the pseudoaxial OTMS group at C4. This cyclo- addition helped to achieve the densely functionalized 2,8-dioxabicyclo[3.2.1]octane Sore structure of the zaragozic acids and prevented the potential problems associatec with the formation of this motif through intramolecular acid—catalyzed ketalizatior of an open-chain 1,3-diol precursor.](https://figures.academia-assets.com/35916932/figure_431.jpg)
![shemicai reaction to obtain enhanced catalytic activity and selectivity | 17]. In this chapter we present the latest developments in cascade reactions catalyzed by heterogeneous nanocatalysts. Tandem transformations mediated by nanocatalysts can be divided into two types of reactions, involving monofunctional and multifunc- tional catalyzed processes (Scheme 8.1) [20]. We highlight the synthesis approach of these novel nanocatalysts, which have been used successfully to produce active, selective, and durable heterogeneous catalysts, and summarize their properties and the cascade reactions which they promote. We then consider potential applications for these new catalvsts and look into the future.](https://figures.academia-assets.com/35916932/figure_432.jpg)


![base. Under optimized conditions, full conversion of iodobenzene to 9 and 10 was attained, giving a 9/10 ratio of 30:70. For 4-methoxyiodobenzene, the conversion was also complete, but the selectivity was lower (50%). No by-products, such as benzene or anisole, were detected in any case. Meanwhile, the catalytic system can be reused without a loss of activity or selectivity. In addition to the use of Pd nanocatalysts for cascade reactions, other metal nano- catalysts were reported by several groups [31]. For example, Sun et al. investigated gold nanoparticles supported on hydroxyapatite as an efficient multifunctional cata- lyst for the rapid and direct synthesis of imines and oximes from amines under mild conditions by a facile tandem oxidation—condensation pathway. The Au/HAP was synthesized through depositing Au nanoparticles onto the as-prepared hydroxyapatite [HAP, Ca,,(PO,),(OH),]. Physical and chemical characterizations demonstrated that the existing Au was in a metallic state with an average diameter of about 3.6nm. To demonstrate the general applicability of the Au/HAP catalyst for direct imine, as well a the scope of the process, various alcohols and amines were investigated (Table 8.3) [32]. All aromatic alcohols reacted with aniline to give the product desired, 11 in excellent yields. Benzyl alcohols with electron-donating groups reacted smoothly, whereas substitution with electron-withdrawing groups on the benzene ring decreased the reactivity. High yields were still generally obtained in the latter case except for with 4-nitrobenzyl alcohol with a strongly electron-withdrawing group. Although a longer reaction time was needed, Au/HAP also displayed high activity in allylic alcohol oxidation. For example, cinnamyl] alcohol was selectively oxidized to the corresponding imine with 95% conversion in 7.5 h. Furthermore, unactivated aliphatic alcohols could also be oxidized smoothly to the corresponding imines in high yield. Sorbitol 13 is considered to be a new-generation green energy platform, replacing syngas, used to produce biofuels, chemicals, and hydrogen [33]. It is generally a eee ee se Pees oP ale lank oe wm ARK ast Lalla ad kes](https://figures.academia-assets.com/35916932/figure_434.jpg)
![TABLE 8.3 Cascade Synthesis of Imines from Various Alcohols and Amines* further hydrogenation. The process, especially hydrolysis under acidic conditions, is not energy efficient and also is not as green as desired [34]. Yan et al. found that Ru nanoparticles could achieve the one-step conversion of cellobiose 12 to C,-alcohols by selectively breaking the C-O—C bonds via selective hydrogenation under 40 bar of H, pressure (Scheme 8.5) [35]. Ru nanoparticles were prepared by reduction of the Ru inorganic salt in the presence of poly(N-vinyl-2-pyrrolidone) in refluxing an ethanol—water solution. A typical micrograph of ruthenium nanoparticles has a narrow unimodal size distribution with a diameter of about 2.4nm. The authors found that increasing the pH from 2 to 7, then to 10, resulted in an acceptable decrease in cellobiose conversion, but caused a dramatic change in product distribution (Table 8.4). Sorbitol was obtained quantitatively under acidic conditions (pH2), indi- cating the occurrence of a two-step process. At pH7, 3-B-p-glucopyranosyl-p- glucitol, the corresponding sugar alcohol of cellobiose, was the main product (64.8% in a conversion of 87.8%), and the selectivity to sorbitol decreased to 26.4%. A small amount of glucose was also observed. ESI-MS and tandem MS analysis evidenced the appearance of dideoxyhexitol (7.2%). This very important product was observed further in basic conditions. Under basic conditions (pH10), the selectivity to 3-B-p- glucopyranosyl-p-glucitol and sorbitol decreased slightly to 79.7%, whereas that for the other C,-alcohols (mainly dideoxyhexitol) increased from 7.2% to 17.1%, demonstrating the precise cleavage of the C,—O in the glycosidic bond in cellobiose.](https://figures.academia-assets.com/35916932/table_002.jpg)


![“Reaction conditions: indole (1.5 mmol), enone (1.0mmol), nano TiO, (10mol%), TMSCN (9.0mmol), anhydrous dichloromethane (3.0 mL), reaction time: 6.0h, reaction done at room temperature. throughout the twentieth century. Among them, the catalytic synthesis of indole ring systems through cycloadditions of 2-haloanilines with alkynes has proven to be the most powerful tool. Generally, this reaction proceeds via an intermediate alkyne 16, which then cyclizes in situ (Scheme 8.6) [40]. Because palladium complexes can catalyze both the Sonogashira cross-coupling reaction and the subsequent ring-closure reaction, they have been employed most frequently for rTABLE 8.5 One-Pot 1,4-1,2 Additions of Indoles to Enones Catalyzed by Nano TiO,“](https://figures.academia-assets.com/35916932/figure_436.jpg)

![one-pot catalytic synthesis of indoles 17. One example of a cascade reaction cata- lyzed by heterogeneous Pd(II) nanocatalysts was reported by Djakovitch et al. They successfully introduced the Pd(II) complex on mesoporous silica [Pd(ID/SBA-15] for one-pot indole synthesis. The Pd(I])/SBA-15 was obtained by grafting the [PdCl, { PPh,(CH,),—SiCH,(OCH,CH,), },] complex via the condensation of ligand alkoxide moieties with surface silanols inside the pores of a calcined SBA-15 mesoporous silica. The hexagonal ordered mesoporous structure of the palladium- functionalized mesoporous material was confirmed by x-ray diffraction analysis, and the integrity of the molecular metal precursor, in particular the phosphine ligand, was indicated by *!P CP-MAS NMR. The authors evaluated the catalytic activity of this Pd(I])/SBA-15 for the synthesis of dual-functionalized indoles. In general, it could obtain high conversions and selectivities, leading to moderate to high isolated yields using 1.0mol% Pd catalyst. However, some of these reactions were found to be quite slow: up to 2 days for the aliphatic substance (Table 8.6). Meanwhile, the Pd(II)/SBA-15 catalysts were found to be recyclable up to five runs, giving generally quantitative conversions.](https://figures.academia-assets.com/35916932/figure_437.jpg)
![SCHEME 8.7 Simplified structure of a monolayer of graphene oxide. Graphene, a one-atom-thick two-dimensional sheet of sp’-hybridized carbon atoms, has had a tremendous impact on many areas of modern chemistry [41]. The use of gra- phene-like catalysts for cascade reactions is a relatively new area with outstanding potential [42]. Recently, Jia et al. revealed the application of graphene oxide (GO) in a multistep-sequence reaction [43]. The simple structure of GO was shown in Scheme 8.7, which revealed that ithas a range of functional groups, including alcohols, epoxides, and carboxylic acids. As a result, GO tended to have highly acidic and strongly oxidizing properties. Based on the physical—chemical properties of GO, they designed one tandem way to form a chalcone compound catalyzed by only a single GO nanomaterial in a single reaction (Scheme 8.8). The catalytic results were summarized in Table 8.7. Both electron-rich (e.g., p-methoxy-substituted) and electron-poor (e.g., p-nitro-substituted) aryl alkynes or methyl ketones were coupled successfully with similarly electron-rich](https://figures.academia-assets.com/35916932/figure_438.jpg)





![SCHEME 8.13 One-step synthesis of styrene carbonate from styrene catalyzed by Au/R201 nanocatalysts. Carbon dioxide fixation has received much attention from the viewpoint of preserva- tion of the Earth, and the reaction of carbon dioxide with epoxides to produce cyclic carbonates has been of great interest as a useful method for the fixation of CO, by chemical processes [53]. Xiang et al. synthesized a strong basic resin R201-supported nanogold catalyst, which was very active for transformation from the styrene to styrene carbonate 24 through two-step epoxidation and cycloaddition reactions (Scheme 8.13). Influence factors such as Au loadings, CO, pressure, temperature of CO, cycloaddition reaction, and CO, addition order were investigated in detail. A styrene carbonate yield of 51% was obtained over 0.01 wt% Au loading of catalyst using a multistep synthetic process. The pronounced cooperative effect of quaternary ammonium cation of the support and nanogold particle resulted in its good performance in the one-pot synthesis of styrene carbonate directly from styrene. The simple one-componentheterogeneous catalyst Au/R201 was readily separated after the reaction and reused without catalyst leaching, which is of great interest as an industrial potential [54].](https://figures.academia-assets.com/35916932/figure_443.jpg)
![3.4 Binary Organometallic-Based Multifunctional Nanocatalysts Among the one-pot multistep sequences, those catalyzed by organometallic complex have reached a pivotal role in synthetic chemistry, owing primarily to the incredible diversity of the transformations available. However, ithas remained largely unexploited in the highly efficient organometallic nanocatalysts with two or more different active sites since the general interference between organometallic complexes. Recently, our complex—were immobilized onto mesoporous silica sequentially, using their corresponding organosilanes in 2-propanol or toluene as the solvent in the first step and toluene as the solvent in the second step (Scheme 8.14). The authors used both MCM-41 and SBA-15 mesoporous silicas as support materials and also investigated the effect of different sequential grafting of the two organosilanes in 2-propanol or toluene on the structures and the catalytic properties of the resulting bifunctional catalysts. By using the resulting amine/Pd(I])—-diamine bifunctional mesoporous material as a catalyst, the occurrence of the Sonogashira and Henry reactions was demonstrated in one pot (Scheme 8.15). Yields of approximately 100% in 2.5h for the Sonogashira reaction and approximately 100% in 45 min for the Henry reaction were obtained in the presence of this bifunctional catalyst when the reactions were run individually. When the bifunctional catalyst was used to catalyze the Sonogashira— Henry reactions in tandem in one pot, a yield of up to approximately 60% of the Sonogashira—Henry product in 5h was obtained.](https://figures.academia-assets.com/35916932/figure_444.jpg)

![Nobel metal nanoparticles supported on metal oxides are among the most important types of heterogeneous catalysts. In general, the metal oxides were considered the support, which might alter the catalytic properties by changing the shape or electronic structure of the metal. However, there are a few examples of using the and metal oxide together as the active site for catalysis [57]. Recently, Yamada et al.](https://figures.academia-assets.com/35916932/figure_446.jpg)


![iminium ion catalyst, with proline-derived chiral pyrrolidine as an excellent enamine catalyst, for a one-pot asymmetric reaction that generated cascade prod- ucts 28 with more than one chiral center. They synthesized two differently func- tionalized star polymers that allowed for the simultaneous use of four catalysts that could not all exist together (Scheme 8.20). These star polymer catalysts could give excellent yields (89%) and stereoselectivity (100: 7 dr, >99% ee for the major diastereomer) of the cascade product in 2 days for a one-pot multiple-component cascade reaction [63].](https://figures.academia-assets.com/35916932/table_007.jpg)




![3.4 CONCLUSIONS AND PERSPECTIVES ae | The three-enzyme-containing polymersome has a particle diameter of about 100 nm. The catalytic activity of this novel nanocalyst was tested by choosing the acetate-protected glucose 30 as substance. External CALB was used to first con- vert the substrate into glucose, which was then used by GO. and HRP to generate ABTSC** (Scheme 8.23). The results showed that the multienzyme could finish this sequence and be removed from the solution by a single filtration step. The kinetic experiment showed that this progress curve of the cascade reaction could be considered a two-enzyme reaction model since HRP did not influence the overall kinetics, which was perhaps as a result of its location on the surface of the polymersome [66].](https://figures.academia-assets.com/35916932/figure_453.jpg)
![SCHEME 9.1 Tandem reaction combining Cu catalysis and Pd catalysis. Later, a similar strategy involving tandem asymmetric conjugate reduction/alkyl- Palladium is a remarkable metal that can serve as either a Lewis acid catalyst or a transitio metal catalyst in organic synthesis and catalysis. With dramatic advances made in th field of Pd-catalyzed cascade reactions, the catalytic cascade or tandem process combinin, palladium and other metals has emerged and demonstrated great potential in searchin: for new reactivity and selectivity. In 2004, Dijk et al. reported a tandem asymmetri conjugate addition/allylic substitution reaction by using combined copper catalysis an palladium catalysis for the total synthesis of natural products (—)-pumiliotoxin [5]. Thi tandem catalysis consisted of a copper salt complex—catalyzed asymmetric conjugat addition and subsequent palladium-catalyzed diastereoselective allylic substitution, pro ducing the chiral building blocks 3 in high reactivity and excellent enantioselectivit (Scheme 9.1). Notably, sequential addition of the two catalysts was essential to avoid th possible interference of two catalysts and to ensure the success of the reaction. Later, a similar strategy involving tandem asymmetric conjugate reduction/alky]](https://figures.academia-assets.com/35916932/figure_454.jpg)





![In 2011, Panteleev et al. reported a domino alkyne arylation and N-arylation pro- cess by combining rhodium and palladium catalysis [10]. Based on a Rh-catalyzed alkyne arylation and subsequent Pd-catalyzed intramolecular N-arylation process, this multicatalytic cascade reaction efficiently incorporated amine-pendant aryl alkynes 25 and aryl boronic acid into functionalized dihydroquinoline derivatives (Scheme 9.7).](https://figures.academia-assets.com/35916932/figure_460.jpg)



![Later, Shimada et al. reported a novel tandem oxidation—reduction reaction with a combination of two fundamentally distinct Ru catalysts [13]. By means of this strategy, the racemic secondary benzylic alcohols could be transformed efficiently into (R)-enantiomers (Scheme 9.10). This catalytic system, containing two different chiral ruthenium catalysts, provides an alternative to chiral secondary alcohol syn- thesis beyond direct reduction or addition protocols.](https://figures.academia-assets.com/35916932/figure_464.jpg)


![SCHEME 9.13 Tandem reaction combining two different imidazolidinone catalysts. eactivity and selectivity control has become an appealing topic. In 2005, Huang et al. reported a tandem asymmetric conjugate reduction-fluorina- ion reaction by an efficient combination of iminium and enamine catalysis using two listinct secondary amine catalysts [16]. This method offered direct access to chiral nultifunctionalized aldehydes from B-substituted enals and electrophilic florinated eagents in a biomimetic way (Scheme 9.13). The diastereoselectivity of the products varied depending on the catalyst combination (Scheme 9.14). The chemistry pre- ented here demonstrated for the first time the power of the multicatalysis process for ontrol of the product diastereoselectivity based on the cycle-specific catalysis ‘oncent. In 2006, Zhao and Cordova reported a similar strategy with a combination of Jérgensen—Hayashi catalyst and (R)-proline [17]. Upon catalysis with the Jorgensen— Hayashi catalyst, the multifunctionalized amino acid derivative desired could be obtained efficiently (Scheme 9.15). While utilizing a catalyst combination com- prising a J@rgensen—Hayashi catalyst and (R)-proline, the diastereoselectivity of the products was inverted (Scheme 9.16). The catalyst combination strategy offers an alternative to selective collection of all the diastereomers of chiral molecules with multiple stereogenic centers.](https://figures.academia-assets.com/35916932/figure_467.jpg)




![In 2008, Chi et al. reported a tandem reaction of indoles, o,$-unsaturated uldehydes, and methyl vinyl ketone (MVK) for the synthesis of chiral indole deriva- ives with two stereogenic centers [19]. To avoid the interference of the two secondary amine catalysts and cocatalyst acid, the soluble star polymer-based site isolation nethod was adopted, whereby the supported imidazolidinone catalyst promoted nitial Friedel-Crafts alkylation and the supported pyrrolidine derivative promoted he following Michael addition to MVK (Scheme 9.19). Notably, simple combination of these catalysts in one pot didn’t mediate the cascade reaction efficiently despite he fact that the MacMillan imidazolidinone and pyrrolidine catalyst can efficiently sromote separate Friedel-Crafts reaction and Michael addition, respectively. Moreover, when the pyrrolidine catalyst was replaced by its enantiomer, a diaste- ‘eomer of the product could be obtained with high enantioselectivity. This study resented a novel solution to the efficient combination of incompatible substrates und catalysts.](https://figures.academia-assets.com/35916932/figure_472.jpg)




![superior choice for reactivity and selectivity control. transformation (Scheme 9.23). The imidazolidione promoted asymmetric Friedel- Crafts alkylation of N-methyl] indole and crotonaldehyde, generating chiral alde- hyde 73, which then participated in the following proline-promoted aza-Michael reaction to dibenzylazocarboxylate delivering the polysubstituted chiral aldehydes 74 (Scheme 9.24). By means of further combination of various nucleophiles and electrophiles, the cycle-specific strategy allowed for implementation of the olefin hydroamination, hydrooxidation, and aminooxidation for the selective synthesis of functionalized molecules with multiple stereocenters. Compared with previous reports involving a single catalyst, this method provides a complementary or superior choice for reactivity and selectivity control. In 2011, Desmarchelier et al. reported a tandem Michael—amination reaction combining secondary and primary amine catalysts based on two distinct enamine catalysts [22], affording synthetically valuable y-nitro aldehydes with a nitrogen- containing quaternary carbon at the o-position (Scheme 9.25). The reaction began with a Jorgensen—Hayashi catalyst-promoted Michael addition of aldehyde to trans- B-nitroalkene, yielding intermediate 76, and a cinchonine-derived primary amine catalyst promoted aza-Michael addition to dibenzylazocarboxylate. This chemistry highlights the versatility of secondary amine catalysis in the activation of simple aldehydes and the complementary activation ability of primary amine catalysts toward sterically hindered o,a-disubstituted aldehydes.](https://figures.academia-assets.com/35916932/figure_477.jpg)





![In 2009, Lathrop and Rovis succeeded in developing a cascade Michael addition/ cross-benzoin reaction by combining chiral iminium ion catalysis and achiral nucle- ophilic carbene catalysis [26]. This relay catalysis involved a consecutive secondary amine-—catalyzed asymmetric Michael addition of active methylene compounds to a, B-unsaturated enals and achiral N-heterocyclic carbene-mediated intramolecular cross-benzoin condensation, affording the enantiomerically enriched polysubstituted cyclopentanones 42 with high reactivity and enantioselectivity (Scheme 9.29). Subsequent control experiments revealed that the two catalysts worked in a coopera- tive manner to promote the two reactions concurrently. In fact, the chiral intermediate aldehyde 41 was susceptible to undergoing retro-Michael in the presence of a Jgrgensen—Hayashi catalyst, hence could erode the yield and selectivity. Again, this chemistry highlighted the power of cascade catalysis over stepwise strategy to promptly utilize the unstable and sensitive intermediate at the next cycle to enhance the synthetic efficiency and selectivity. Later, the same group expanded this chemistry further by developing a cascade Michael addition/cross-benzoin condensation sequence of enolizable aldehydes 43 and activated enones 44 [27]. The reaction proceeded by means of enamine activation of aliphatic aldehydes to induce an asymmetric Michael addition to activated enones followed by an intramolecular cross-benzoin condensation (Scheme 9.30). Compared with their previous work, complex cyclopentanones with complementary substitution patterns were observed. Screening of the reaction parameters revealed that the chiral triazolium catalyst was necessary to ensure a satisfactory stereochemical outcome. Further mechanistic insights indicated that the high diasteroselectivity observed attributed to the secondary amine—induced epimerizing of the o-position of intermediate aldehyde 89.](https://figures.academia-assets.com/35916932/figure_483.jpg)


![Incorporation of the new catalysis concept and N-heterocyclic carbene catalysis into multicatalytic systems emerged as a new direction to build up a complex scaffold such as those often found in natural products. Liu et al. developed an enantioselective Diels—Alder reaction via trienamine catalysis of indole-2,3-quinodimethane and activated alkenes. More recently, they combined this Diels—Alder process with N-heterocyclic carbene catalysis for the rapid generation of chiral cyclopentanone- fused tetrahydrocarbazoles with diverse substitution [30]. Mechanistically, with the N“HCLCTOCYCHC CalrOChe Caldlyst. B-Ketophenyltetrazolesulfones are readily available nucleophiles in the facile intr luction of alkenyl and alkynyl moieties into organic molecules. By means of a simil: strategy presented by Rovis and Enders, Jérgensen’s group achieved the efficie -onstruction of chiral cyclopent-2-one derivatives via a tandem Michael addition/cros yenzoin/Smiles rearrangement sequence [29]. Secondary amine—promoted Micha iddition of B-ketophenyltetrazolesulfone to o1,8-unsaturated enal delivered 1,5-dica yonyl compounds as suitable precursors for N-heterocyclic carbine—promoted intram: ecular cross-benzoin coupling (Scheme 9.32). Upon the formation of benzoin product yase-mediated Smiles rearrangement of the o-hydroxyl phenyltetrazolesulfones ga\ iccess to 2,4-disubstituted cyclopent-2-ones. Considering the prevalence and importanc of a transformable enone moiety in natural products and organic synthesis, this approac »ffers a general and practical strategy toward the assembly of these valuable targets.](https://figures.academia-assets.com/35916932/figure_486.jpg)




![One year later, Rahaman et al. reported a cascade reaction of aldehyde and nitro alkanes for the synthesis of functionalized y-nitro aldehydes with high reactivity [33]. Generation of the intermediate nitro alkenes involved cooperative secondary amine/hydrogen bonding—promoted condensation of nitromethane and aldehydes. Consequently, the newly formed nitro alkenes promptly participated in the next cycle cocatalyzed by the two catalysts (Scheme 9.37). In 2011, Lin et al. reported a combination of hydrogen-bonding catalysis and iminium catalysis and its application in a cascade [3+2] cycloaddition process, which gave rise to substituted chiral pyrrolidines with high stereoinduction [34]. A relatively weak acidic hydrogen-bonding donor catalyst efficiently promoted the formation of reactive intermediates 114, which immediately took part in the follow- ing [3+2] cycloaddition process (Scheme 9.38). Further study revealed that intro- duction of the hydrogen-bonding donor catalyst was also crucial to tune the selectivity of the products.](https://figures.academia-assets.com/35916932/figure_491.jpg)

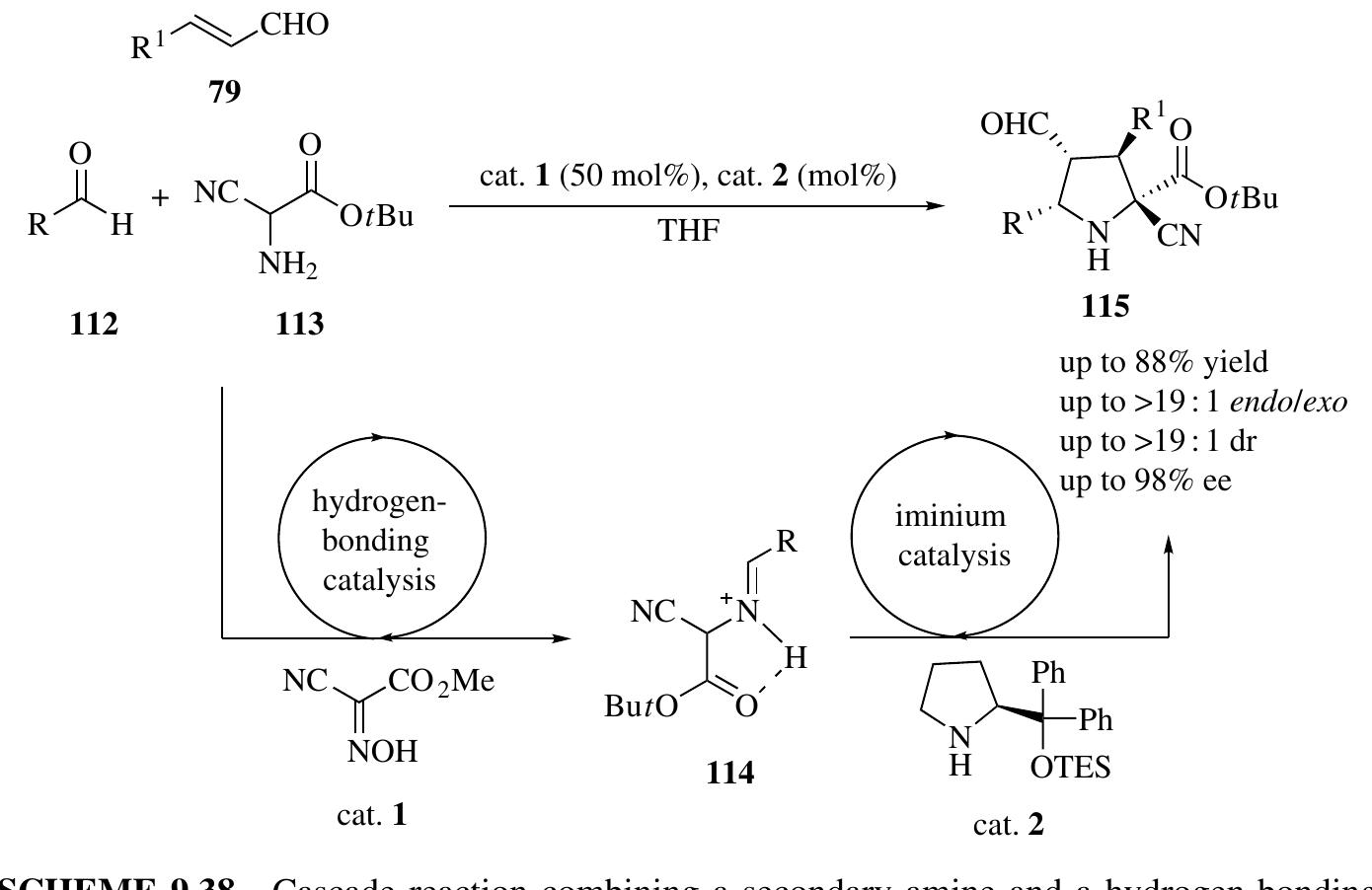


![these robust catalysts in cascade reaction is largely undeveloped. In 2010, Fillow et al. discovered a cascade Michael—Stetter reaction by a combination of achira nucleophilic tertiary amine catalysis and chiral N-heterocyclic carbene catalysis [39] Mechanistically, the cascade reaction proceeds via a tertiary amine—promotec intermolecular oxa-Michael addition and a following N-heterocyclic carbene- mediated intramolecular Setter reaction. Impressively, the different activation mod and substrate recognition ability of the two nucleophilic catalysts were combinec successfully to cooperatively couple the electron-deficient alkynes and salicylalde hydes, providing straightforward access to chiral benzofuranones with a quaternar stereogenic center (Scheme 9.43). Clana Tix barantnce nana eernekow OOF? BDeanetad kaos. Dennctan amen kefwwantanwns](https://figures.academia-assets.com/35916932/figure_496.jpg)



![SCHEME 9.46 Cascade reaction combining (S)-proline and an AgOTf catalyst. Udldlyst, HdVe VOC GIsC10sed, In 2007, Ding and Wu reported a cascade reaction combining enamine and Ag catalysis for the synthesis of 1,2-dihydroisoquinoline derivatives [42]. Initially, the nucleophilic enamine derived from the ketone and proline attacked the imine that arose from the aldehyde and the amine, forming the Mannich base 140, which then participated in a hydroamination reaction toward pendant alkyne to build the final product (Scheme 9.46).](https://figures.academia-assets.com/35916932/figure_500.jpg)








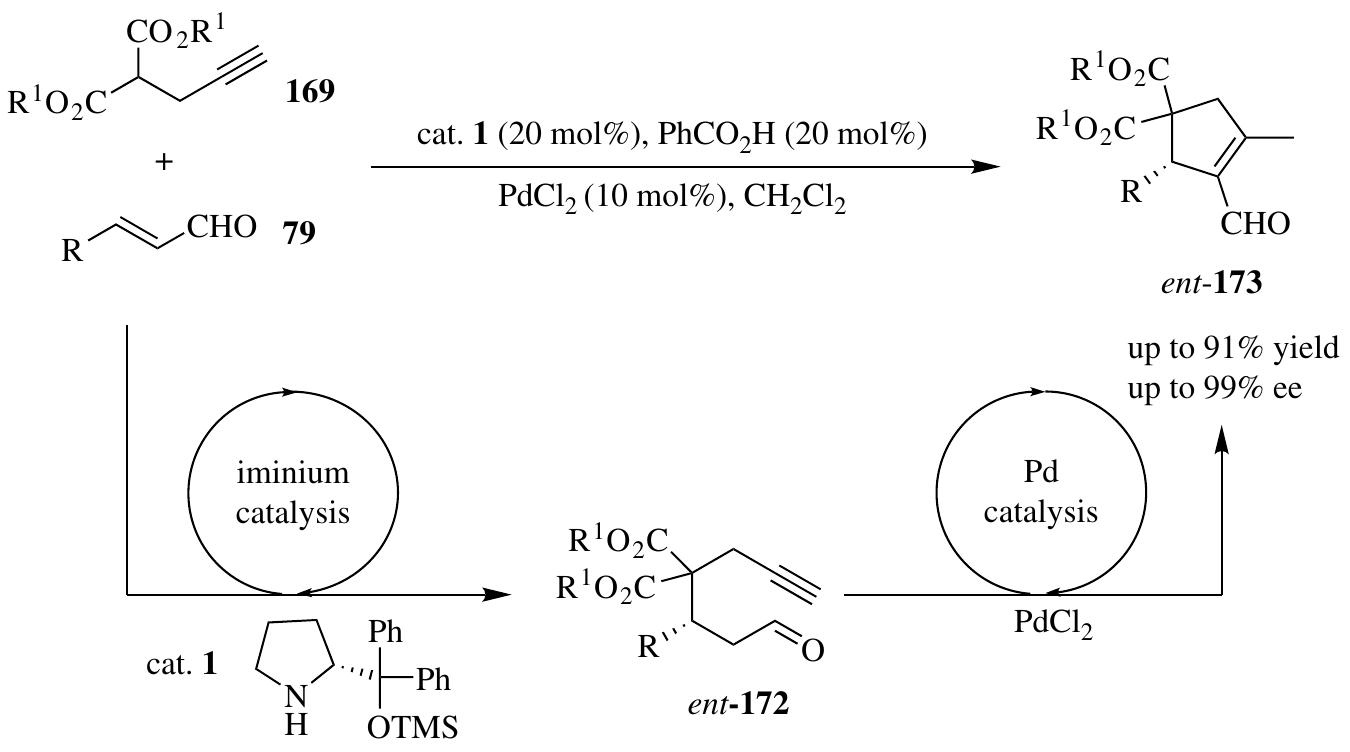

![SCHEME 9.57 Cascade reaction combining a secondary amine and a Cu or Au catalyst. Jensen et al. also displayed their efforts toward this direction by combining an Au(I) complex and a secondary amine catalyst. They first demonstrated an enone- nvolved Michael addition/cycloisomerization process by means of primary amine ind Au catalysis [51]. They then embarked on a similar reaction by replacing enone with enals, while the catalytic system comprised a secondary amine and a Cu or Au catalyst [52], which yielded the cyclic chiral enal 176 with comparable results ‘Scheme 9.57). Recently, Lin et al. demonstrated that the propargyl alcohol could participate in such a transformation for the synthesis of chiral dihydrofurans [53]. The reaction began with a challenging oxa-Michael addition to cinnamaldehyde derivatives, which was followed by a secondary amine/Pd complex—catalyzed nucleophilic addition/ isomerization of the alkyne moiety in excellent yields and enantioselectivities (Scheme 9.58). Since the oxa-Michael addition of propargyl alcohol to o,B- unsaturated aldehydes was a slow process, this cascade reaction proceeded through a dynamic kinetic asymmetric transformation (DYKAT) process, whereby it made the overall reaction proceed efficiently and with high stereocontrol using the second reaction with precise stereocontrol to shift the first reversible oxa-Michael addition selectively.](https://figures.academia-assets.com/35916932/figure_511.jpg)


![Considering the rapid growth of asymmetric construction of oxindoles, Sun et al. recently reported their assembly of chiral spirooxindoles by combining secondary amine and palladium catalysis in a cascade reaction [55]. The reaction was initiated by the reversible Michael addition of 3-substituted oxindole to enal, which was followed by a metal/organic-cocatalyzed carbocyclization of the alkyne tether (Scheme 9.60). Similar to the aforementioned dynamic kinetic asymmetric transformations, this chemistry highlighted the cooperative effects of the two catalysts in the same reaction vessel, while either catalyst could not solely promote the overall reaction, and unsat- isfactory results were observed when this reaction was conducted in a two-step mode.](https://figures.academia-assets.com/35916932/figure_514.jpg)








![SCHEME 9.67 Cascade reaction combining Au and phosphoric acid catalysis. Almost at the same time, Liu and Che published a cascade intermolecular hydroamination/asymmetric reduction sequence, which included achiral Au complex— catalyzed hydroamination of aryl amines and chiral phosphoric acid—promoted Hantzsch ester reduction to afford secondary aryl amines [70]. More recently, the same group reported a tandem one-pot assembly of functionalized tetrahydroquino- lines from amino aldehyde and alkynes by combining Au and chiral phosphoric acid catalysis [71]. The reaction was initiated by Au-promoted quinololine 210 generation, followed by an enantioselective HEH-incorporated transfer hydrogenation process (Scheme 9.67). Chiral Brgnsted acid—promoted Pictet-Spengler-type reaction of electron-rich alkenes with iminium ion species was prevalent in asymmetric synthesis. Incorporation of this reaction into a cascade process will undoubtedly provide a straightforward and powerful strategy for the construction of complex structures. In 2009, Muratore et al. reported a tandem reaction combining achiral Au and chiral phosphoric acid catalysis for the synthesis of polycyclic indole architec- tures [72]. The cascade sequence began with an Au(I)-catalyzed cycloisomeriza- tion of substituted alkynoic acid to form enol lactones 214, which immediately underwent ring opening upon exposure to hard tryptamines and an acid promoter to give rise to Pictet-Sepengler reaction precusors. Finally, under the influence of chiral phosphoric acid, dehydrative cyclization via N-acyliminium ion inter- mediates afforded the product desired (Scheme 9.68). Impressively, although the reaction was performed at relatively high temperatures (80°C for 24h and then 110°C for 24h), satisfactory enantiocontrol of this cascade reaction was still achieved.](https://figures.academia-assets.com/35916932/figure_523.jpg)






![reaction to assemble biologically valuable tetrahydrocarbazoles [79] (Scheme 9.74). First, Friedel-Crafts alkylation of indoles to aliphatic nitroalkene with a iodine-sub- stituted pendant produced C3 functionalized chiral indole 235, which was susceptible to take part in a second Friedel-Crafts alkylation under the influence of soft Lewis acidic AgSbF, and a subsequent 1 ,2-shift (Ciamician—Plancher rearrangement) to lib- erate the annulated products desired.](https://figures.academia-assets.com/35916932/figure_530.jpg)


